
Cleanroom Standards for Cell Therapy Manufacturing Guide
Cell therapies cannot be terminally sterilised — every contamination event in the manufacturing environment destroys the batch and, for autologous therapies, the only batch that exists for that patient. For pharmaceutical manufacturers, biotech startups, and hospitals building CAR-T capacity in Canada, cleanroom standards for cell therapy manufacturing are the difference between regulatory approval and a complete shutdown. This guide covers every framework you need: ISO 14644, FDA cGMP 21 CFR Part 211, and EU GMP Annex 1 — applied specifically to cell therapy.

- Three frameworks govern cell therapy cleanrooms: ISO 14644 (particle classification), FDA cGMP 21 CFR Part 211 (US), EU GMP Annex 1 (global gold standard)
- Grade A / ISO 5 is mandatory for all critical aseptic steps — CAR-T viral transduction, final formulation, fill-finish, sterile connections
- Closed-system cell expansion can run in Grade C (ISO 7) or Grade D — the biggest cost lever in cell therapy facility design
- IQ/OQ/PQ validation + re-certification every 6–12 months is mandatory under ISO 14644-2, not optional
- Modular GMP cleanrooms deliver validated cell therapy suites in 8–16 weeks — 30–40% faster and cheaper than conventional construction
Why Cleanroom Standards Are Non-Negotiable in Cell Therapy
Unlike small-molecule drugs or even conventional biologics, cell therapies cannot be terminally sterilised. Living cells cannot be heat-treated, radiation-sterilised, or subjected to final filtration without destroying the therapeutic product. Every particle, microorganism, or cross-contamination event in the manufacturing environment is a direct patient safety risk.
Three factors make cell therapy cleanroom compliance significantly harder than standard sterile drug manufacturing:
- Extended open processing windows — cell expansion runs 7–14 days in bioreactors and open culture systems, creating prolonged contamination exposure compared to minutes in conventional fill-finish operations
- Patient-specific autologous batches — for CAR-T therapies, each batch is a single patient’s material. Cross-contamination between batches can be life-threatening and constitutes a critical GMP failure mode that cannot be remediated
- No post-process sterilisation option — temperature excursions, microbial contamination, and particulate exposure directly compromise cell viability and therapeutic potency with no salvage option
For Canadian facilities, regulatory consequences are severe: Health Canada enforcement actions, FDA Warning Letters for US-market products, mandatory facility shutdowns, and product recalls that permanently damage regulatory standing. Getting the cleanroom classification strategy right before construction begins is not optional — retrofitting a facility after an inspection failure costs far more than building correctly the first time.

The Three Regulatory Frameworks That Govern Cell Therapy Cleanrooms
1. ISO 14644 — The Technical Classification System
ISO 14644-1:2015 defines cleanroom classification by maximum allowable airborne particle concentrations per cubic metre at ≥0.5 μm. The standard covers ISO Class 1 (ultra-clean semiconductor fabs) through ISO Class 9. Cell therapy manufacturing uses ISO Classes 5 through 8 depending on the process step and exposure risk.
ISO 14644-2 (2015) adds the monitoring and re-qualification schedule that underpins ongoing GMP compliance. It mandates re-qualification every 6 months for ISO Class 5 environments and every 12 months for ISO Class 7–8, plus continuous particle monitoring during all critical aseptic operations. ISO 14644 defines the measurement methodology and particle limits — it does not specify which classification applies to each manufacturing step. That comes from the GMP frameworks below.
2. FDA cGMP — 21 CFR Part 211 and Cell Therapy Guidance
FDA 21 CFR Part 211, Section 211.42 sets design and construction requirements for aseptic processing areas. The FDA does not reference ISO classes directly — instead requiring “appropriate” environmental controls, with industry practice aligning to ISO 14644. Two FDA guidance documents are essential for cell therapy facilities:
- FDA 2004 Aseptic Processing Guidance — defines the “Critical Area” (Grade A / ISO 5) standard, 10–15 Pa pressure differentials, unidirectional HEPA-filtered airflow, and media fill validation requirements
- FDA 2020 Gene Therapy Products Guidance — permits conditional release for short-shelf-life autologous cell therapies based on rapid in-process sterility testing (48–72 hours pre-harvest) rather than 14-day compendial tests
For Canadian facilities, Health Canada GUI-0001 GMP Guidelines align closely with FDA cGMP and reference the same ISO 14644 classification framework. Both apply to compounding pharmacies, biologics manufacturers, and commercial cell therapy producers operating under a Drug Establishment Licence.
3. EU GMP Annex 1 (2022 Revision) — The Global Gold Standard
The 2022 revision of EU GMP Annex 1 (effective August 2023) is the most comprehensive and prescriptive sterile manufacturing standard in the world. Any Canadian or US facility targeting European market approval must be designed to Annex 1 from day one. Three major 2022 changes affect cell therapy facility design:
- Mandatory Contamination Control Strategy (CCS) — a living document that maps every contamination risk across the facility, defines specific control measures at each critical point, and must be maintained throughout the product lifecycle
- Continuous viable monitoring in Grade A — periodic air sampling alone no longer satisfies the requirement; real-time monitoring during all critical operations is mandatory
- Minimum 10 Pa pressure differentials with automated BMS alarming — manual spot checks are no longer sufficient for compliance

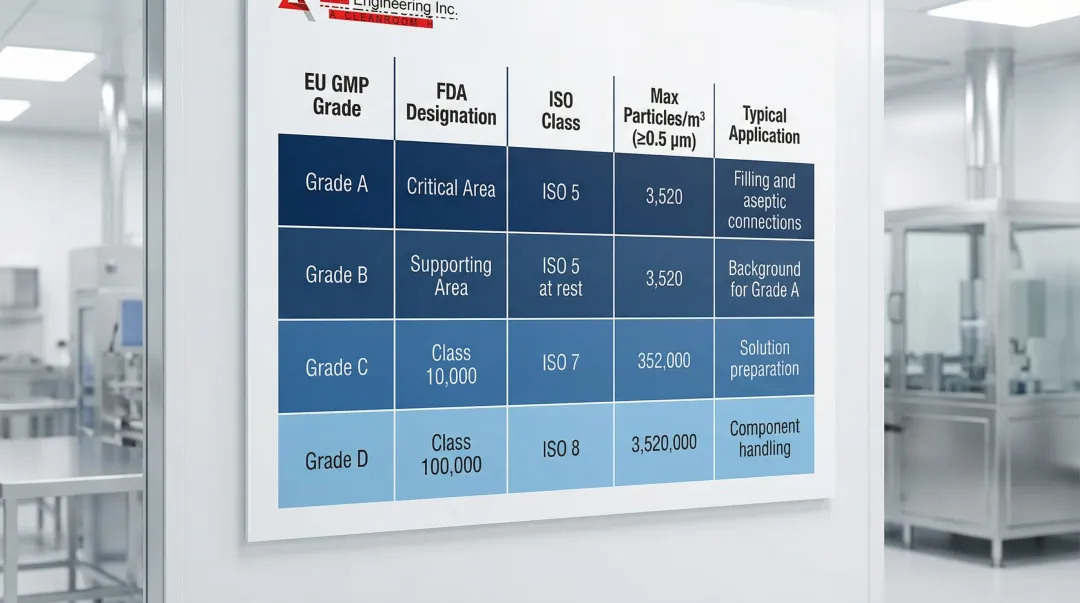
ISO 14644 / EU GMP Grade Classification for Cell Therapy
The table below maps EU GMP grades to ISO classes, particle limits, and specific cell therapy applications. This is the foundation of every cell therapy facility design decision.
| EU GMP Grade | ISO Class | FDA Designation | Max Particles/m³ (≥0.5 μm) | Cell Therapy Application |
|---|---|---|---|---|
| Grade A | ISO 5 | Critical Area | 3,520 | Final formulation, fill-finish, sterile connections, viral vector addition (open), cryopreservation bag loading |
| Grade B | ISO 5 (at rest) / ISO 7 (operation) | Supporting Area | 3,520 / 352,000 | Background environment surrounding all Grade A aseptic operations; gowning area for Grade A access |
| Grade C | ISO 7 | Class 10,000 | 352,000 | Cell isolation, closed-system expansion, media and buffer preparation, secondary gowning |
| Grade D | ISO 8 | Class 100,000 | 3,520,000 | Component staging, equipment preparation, closed bioreactor operation, support areas |
Pressure Cascade — Protecting Classification Integrity
A correctly designed pressure cascade maintains positive pressure differentials from higher to lower classification zones, physically preventing contamination migration. The standard cascade for a cell therapy facility is:
- Grade A: +25 Pa relative to atmospheric
- Grade B: +15 Pa
- Grade C: +10 Pa
- Grade D: +5 Pa
- Unclassified corridor: 0 Pa (atmospheric reference)
When this cascade breaks down — through HVAC failure, door sequencing errors, or pressure sensor drift — contamination travels upstream into critical zones. EU GMP Annex 1 (2022) requires continuous BMS-integrated pressure differential monitoring with automated alarming. Manual periodic checks no longer satisfy the requirement.

The Closed Systems Cost Strategy
Both FDA CAR-T guidance and EU GMP Annex 1 explicitly permit Grade D (ISO 8) backgrounds for processes conducted in fully validated closed systems. This is one of the most impactful cost levers in cell therapy facility design.
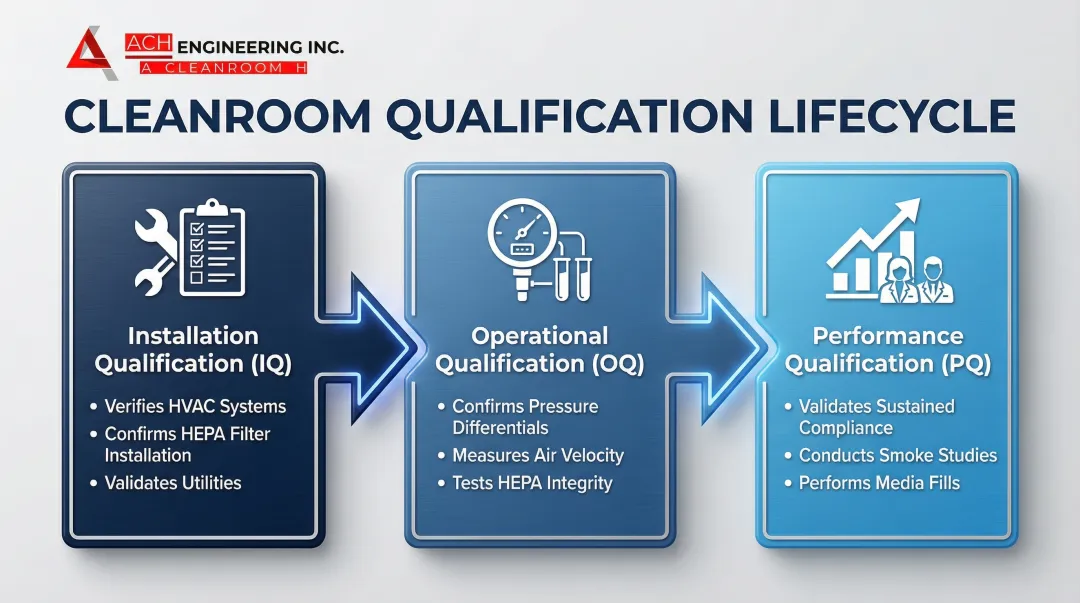
IQ/OQ/PQ — Validation Framework for Cell Therapy GMP Cleanrooms
Commissioning, Qualification, and Validation (CQV) is the structured process that converts a constructed cleanroom into a regulatory-approved manufacturing environment. Every qualification phase generates documentation that forms part of your Health Canada, FDA, or EMA submission package.

Environmental Monitoring Programme Requirements
An Environmental Monitoring Programme (EMP) is continuous surveillance of your cleanroom for both non-viable particles (particle counters) and viable organisms — bacteria and fungi measured via active air samplers, settle plates, and contact plates.
Grade A Monitoring Requirements
- Continuous non-viable particle monitoring at ≥0.5 μm and ≥5 μm during every critical operation — not just start and end of each processing run
- Active viable air sampling at defined locations throughout each processing session
- Settle plates exposed for the entire duration of critical operations
- Contact plates for gloved hands and equipment surfaces after operations
- Zero tolerance: any Grade A viable action limit exceedance triggers product quarantine and mandatory root cause investigation before resuming operations
Grades B–D Monitoring
Monitoring frequency for Grade B–D zones is risk-based under EU GMP Annex 1 and must be defined in the CCS. Typical programmes include viable air sampling each processing shift, surface monitoring at the end of each production day, and non-viable particle monitoring at scheduled intervals. All excursions from alert limits require documented investigation; action limit exceedances require a formal CAPA process before manufacturing can resume.
Modular Cleanroom Construction for Cell Therapy Facilities
Traditional stick-built cleanroom construction is slow, expensive to modify, and difficult to reconfigure as cell therapy processes scale from Phase I clinical to commercial manufacturing. Modular GMP cleanroom construction — prefabricated panels, integrated walkable ceilings, and flush-frame GMP door systems — is now the preferred approach for cell therapy facility design.

The four advantages of modular construction for cell therapy facilities:
- Speed: Modular fit-outs deliver in 8–16 weeks versus 24–40+ weeks for conventional construction — critical when clinical trial timelines and manufacturing licensing cannot wait
- Regulatory documentation: GMP-grade prefabricated modular wall panels meet cGMP surface, joint, and penetration requirements from the factory, reducing IQ documentation burden significantly
- Flexibility: Classification zones can be reconfigured without demolition as manufacturing processes evolve from small-batch clinical to commercial scale — a critical capability for cell therapy facilities where process changes are frequent
- Cost: Modular construction typically delivers equivalent GMP-classified space at 30–40% lower cost than traditional build-out, with lower long-term maintenance costs for coving and surface joints
Building a Cell Therapy or Pharmaceutical Cleanroom in Canada?
ACH Solutions designs and builds validated modular GMP cleanrooms for pharmaceutical, biotech, and cell therapy manufacturers across Canada and the USA. IQ/OQ/PQ ready. 8–16 week delivery. 30–40% lower cost than conventional construction.
Get a Project Quote →Common Compliance Failures in Cell Therapy Cleanrooms
Over-Classifying the Entire Facility
Classifying an entire facility to Grade B when only targeted zones require it is one of the most expensive mistakes in cleanroom facility design. Every square metre of Grade B space demands higher-specification HVAC, more extensive gowning protocols, additional monitoring points, and greater ongoing operational cost. Risk-based zoning — Grade A only where product is exposed, Grade C for closed-system processing, Grade D for support areas — cuts facility operating costs by 40% or more without compromising product safety or regulatory compliance.
Treating Initial Qualification as Permanent Compliance
A cleanroom that passes IQ/OQ/PQ at commissioning does not remain compliant indefinitely. Classification degrades through HEPA filter loading and seal degradation, HVAC system wear, panel joint deterioration, and changes to occupancy or equipment loading. Facilities relying on initial qualification without a disciplined rolling re-qualification schedule routinely fail FDA inspections and EMA audits. The cost of a regulatory shutdown — lost batches, facility downtime, consultant fees, and the regulatory remediation process — far exceeds the cost of a maintained re-qualification programme.
Applying Batch Frameworks to Personalised Therapies
Standard GMP was written for large-batch conventional pharmaceutical manufacturing, not for autologous cell therapies with 72-hour shelf lives and one-patient batch sizes. Three recurring friction points:
- Sterility testing timelines: 14-day compendial sterility tests exceed most cell therapy shelf lives. FDA permits conditional release based on rapid in-process microbial detection (48–72 hour pre-harvest rapid testing)
- Segregation at scale: Autologous segregation requirements strain facility capacity as patient volumes grow. Dedicated processing suites or validated closed systems are required for commercial throughput
- Traceability systems: Batch record structures designed for multi-unit production need fundamental redesign for single-patient traceability chains that comply with 21 CFR Part 11 and EU Annex 11 electronic records requirements

Conclusion: Get the Classification Strategy Right Before You Build
Cleanroom classification strategy for cell therapy manufacturing is fundamentally a risk analysis problem, not a construction problem. The correct approach starts with your manufacturing process, maps product exposure risk at each step, assigns the minimum classification that manages that risk, and then uses closed systems and modular construction to minimise the high-grade footprint.
The three frameworks — ISO 14644, FDA cGMP, and EU GMP Annex 1 — converge more than they conflict. A facility designed from first principles to satisfy EU Annex 1 will simultaneously satisfy FDA cGMP and ISO 14644. The 2022 Annex 1 revision’s risk-based approach is not at odds with FDA’s flexible guidance — both ask the same question: have you identified every contamination risk and verified a control is in place?
Modular construction makes this practical for cell therapy development timelines. Targeted classification zones, BMS-integrated monitoring, IQ/OQ/PQ-ready documentation, and reconfigurable floor plans can all be delivered in 8–16 weeks — fast enough to meet clinical trial timelines with the regulatory infrastructure that commercial-scale manufacturing submissions require. Contact ACH Solutions to discuss your cell therapy facility requirements.
Frequently Asked Questions
Related Solutions & Resources
GET IN TOUCH
Complete the form below to get in touch with our team.