
Introduction: ISO 13485 and Cleanroom Manufacturing
Many medical device manufacturers mistakenly assume that ISO 13485 certification automatically defines their cleanroom requirements — a costly misconception that can lead to non-conformances during audits. ISO 13485 establishes quality management system expectations but doesn't specify cleanroom classifications—those come from separate standards like ISO 14644.
Cleanrooms remain essential for manufacturers producing sterile or contamination-sensitive devices. The connection lies in ISO 13485's Clause 6.4 (Work Environment), which requires manufacturers to determine and manage environmental conditions that could affect product quality.
That risk-based approach to environmental controls carries significant regulatory weight across North America. The FDA's Quality Management System Regulation (QMSR) incorporates ISO 13485:2016 by reference effective February 2026, while Health Canada's Medical Device Regulations already align closely with the standard — making Clause 6.4 compliance a cross-border priority.
What follows covers how ISO 13485 and cleanroom standards intersect in practice — from classifications and design requirements to validation protocols and implementation strategies.
TLDR: Key Takeaways
- ISO 13485 mandates suitable manufacturing environments but references ISO 14644 for specific cleanroom classifications
- Medical device cleanrooms typically range from ISO Class 5–8 based on device risk and sterility requirements
- Compliant design requires HVAC systems, minimum 10 Pascal pressure differentials, and validated environmental monitoring
- Modular cleanroom builds reduce time-to-compliance and allow for future expansion without full reconstruction
Understanding ISO 13485: Quality Management for Medical Devices
ISO 13485:2016 defines quality management system requirements for medical device manufacturers, covering everything from design controls to production environments. It's the benchmark regulators and customers worldwide use to verify that devices are consistently safe and effective.
Global Regulatory Recognition
The standard's reach expanded with the FDA's QMSR final rule, which amends 21 CFR Part 820 to incorporate ISO 13485:2016 by reference. This brought US requirements into alignment with the EU Medical Device Regulation (MDR).
Health Canada also aligns with ISO 13485, making it the shared foundation for medical device quality management across North America and Europe.
Clause 6.4: Work Environment Requirements
ISO 13485 addresses cleanroom controls through Clause 6.4, which requires:
- Documented work environment requirements where conditions could adversely affect product quality
- Contamination controls (Clause 6.4.2) for sterile device assembly or packaging, including arrangements for controlling microorganisms and particulate matter
- Referenced technical standards — ISO 14644 and ISO 14698 — for cleanroom classification and biocontamination monitoring guidance
Notably, ISO 13485 doesn't prescribe specific cleanroom classes. Manufacturers must determine appropriate environmental controls based on product risk, contamination sensitivity, and what regulators expect for their device type.
The Relationship Between ISO 13485 and Cleanroom Standards
ISO 13485 provides the quality management framework, while technical standards like ISO 14644 define specific environmental parameters.
Regulatory Framework Integration
Three major regulatory frameworks shape how ISO 13485 intersects with cleanroom requirements in practice:
- FDA 21 CFR Part 820 mandates a documented QMS compliant with ISO 13485, covering facilities and controls for device design, manufacture, and packaging.
- EU GMP Annex 1 governs cleanroom grades (A–D) and monitoring for sterile products. ISPE guidance specifies a minimum 10 Pascal pressure differential between adjacent rooms of different grades.
- Health Canada requires environmental controls aligned with ISO 13485 and applicable cleanroom standards, determined by device classification.

These frameworks converge during audits, where compliance gaps across any one of them can affect certification status.
Audit and Validation Expectations
Regulatory auditors and notified bodies verify that environmental control systems are both validated and actively monitored. Where process outputs — such as air cleanliness — cannot be confirmed through monitoring alone, the control system itself must be validated and maintained to confirm it is functioning as designed.
Cleanroom Classification Standards: ISO 14644 and EU GMP Grades
ISO 13485 doesn't specify cleanroom classes directly — it requires manufacturers to define and control their environment based on product risk. ISO 14644-1 provides the classification framework that makes this possible, defining ISO Classes 1–9 by particle concentration per cubic meter. Identifying the right class for your process is the first step toward a compliant controlled environment.
ISO Classification Table
| ISO Class | Particles ≥0.3 µm/m³ | Particles ≥0.5 µm/m³ | Particles ≥5.0 µm/m³ | Typical Application |
|---|---|---|---|---|
| ISO 5 | 10,200 | 3,520 | 29 | Critical aseptic processing |
| ISO 6 | 102,000 | 35,200 | 293 | Sterile device manufacturing |
| ISO 7 | 1,000,000 | 352,000 | 2,930 | General device assembly |
| ISO 8 | — | 3,520,000 | 29,300 | Non-sterile controlled environments |
EU GMP Grades vs. ISO Classes
For sterile medical device manufacturing, EU GMP Grades map directly onto ISO classes — but the correspondence applies to the operational state, not at-rest conditions. Understanding this distinction matters: a room that qualifies at ISO 5 when empty must still meet that threshold during active production. The table below shows how grades align and what airflow each demands.
| EU GMP Grade | ISO Equivalent (Operational) | Airflow Requirement | Application |
|---|---|---|---|
| Grade A | ISO 5 | Unidirectional, 0.45 m/s ±20% | High-risk operations (filling, critical assembly) |
| Grade B | ISO 5–7 | Turbulent | Background for Grade A |
| Grade C | ISO 7–8 | Turbulent | Less critical manufacturing stages |
| Grade D | ISO 8 | Turbulent | Lowest grade for sterile manufacturing |
At-Rest vs. Operational State
Cleanrooms are evaluated under two conditions, and both matter for certification:
- At-rest — Equipment installed, no personnel present. This baseline state is used during initial qualification to confirm the room meets its design class.
- Operational — Normal production running, with personnel and equipment active. Compliance must hold under these conditions throughout manufacturing, not just during qualification.
Medical Device Risk Classification and Cleanroom Requirements
Device risk classification directly determines cleanroom requirements under ISO 13485's risk management framework. Higher-risk devices demand tighter environmental controls — from particle counts to microbial monitoring — while lower-risk devices may only need a controlled, non-classified space.
Risk-Based Environmental Controls
Class III — Implantable Devices (pacemakers, heart valves):
- ISO Class 5-7 for critical assembly steps
- Sterile manufacturing environments
- Stringent environmental monitoring
Class II — Moderate Risk Devices (infusion pumps, surgical instruments):
- ISO Class 7-8 for assembly
- Controlled but not necessarily sterile environments
- Risk assessment determines specific requirements
Class I — Lower Risk Devices:
- ISO Class 8 or controlled (non-classified) environments
- Requirements based on contamination sensitivity
- Clean but not necessarily cleanroom conditions
Sterile vs. Non-Sterile Manufacturing
The sterile/non-sterile distinction is one of the most consequential factors in setting cleanroom classification:
- Sterile devices: Require ISO Class 5-7 environments, validated sterilization processes, and monitoring for both particles and microorganisms
- Non-sterile devices: Typically require ISO Class 7-8 or a controlled environment, with the specific classification determined by contamination sensitivity and intended use

Risk assessment under ISO 13485 determines the required cleanroom classification for both categories.
Cleanroom Design and Construction Requirements for ISO 13485 Compliance
ISO 13485 compliance depends on how well your cleanroom's physical systems — airflow, surfaces, pressure, and material transfer — are designed from the ground up. Each element works together; a gap in any one area can compromise your classification and your audit outcomes.
HVAC and Air Change Rates
Air Change Rates (ACH) are critical for contamination dilution and removal:
| ISO Class | Typical ACH Range | HEPA Filtration | Notes |
|---|---|---|---|
| ISO 5 | 240-480 | 99.97% at 0.3 µm | Unidirectional flow preferred |
| ISO 6 | ~120 | 99.97% at 0.3 µm | Turbulent mixing |
| ISO 7 | 30-60 | 99.97% at 0.3 µm | Most common for device assembly |
| ISO 8 | ~20 | 99.97% at 0.3 µm | Controlled environment |
Research from ISPE confirms that ISO 7 cleanrooms operate effectively at 30-60 ACH when properly designed.
Once airflow rates are established, pressure management becomes the next layer of contamination control.
Pressure Differential Requirements
Positive pressure cascades prevent contamination migration from less clean to cleaner areas:
- Minimum differential: 10 Pascals between adjacent rooms of different grades
- Monitoring: Continuous monitoring with frequent recording throughout production shifts
- Alert levels: Dual approach (alert and action levels) recommended for early warning
Wall, Ceiling, and Flooring Systems
Surface materials must be smooth, non-shedding, and cleanable — properties that affect both contamination control and audit readiness. Common wall panel options include:
- High Pressure Laminate (HPL) panels
- Powder-coated galvanized steel
- uPVC (rigid PVC) panels
Ceilings should use walkable systems with integrated HEPA filters, reducing contamination risks from overhead access during maintenance.
Flooring requires seamless installation to prevent particle accumulation at joints:
- Epoxy coatings
- Vinyl sheet flooring
- Seamless wall coving connections for cGMP compliance
Doors and Material Transfer
Door requirements:
- Flush design to minimize particle generation
- Interlocking systems to prevent simultaneous opening
- Pressure-rated construction
Pass-throughs: Material transfer systems with interlocks, UV sterilization (where appropriate), and protocols to maintain pressure differentials during transfers.
Environmental Monitoring and Validation Requirements
ISO 14644-2 and regulatory guidelines require structured monitoring programs to confirm ongoing cleanroom performance — and under ISO 13485, gaps in that monitoring can directly jeopardize product quality and audit outcomes.
Monitoring Plan Requirements
A risk-based monitoring plan must include:
Continuous monitoring of:
- Particle counts at critical locations
- Pressure differentials between zones
- Temperature and humidity
Periodic monitoring of:
- Airflow velocity and patterns
- HEPA filter integrity
- Microbial contamination (for sterile environments)
Alert and action levels are a key component of any monitoring plan. ISO 14644-2 requires at minimum a single action level, though dual levels — alert and action — provide earlier warning of deviations before they become compliance issues.
Validation Lifecycle: IQ/OQ/PQ
Monitoring alone isn't sufficient; the cleanroom itself must be formally qualified before and during production. This is where the IQ/OQ/PQ lifecycle comes in.
| Stage | Purpose |
|---|---|
| Installation Qualification (IQ) | Verifies equipment and systems are installed per specifications and manufacturer requirements |
| Operational Qualification (OQ) | Confirms systems operate within specified ranges under varying conditions |
| Performance Qualification (PQ) | Demonstrates the cleanroom consistently meets specifications under real production conditions |
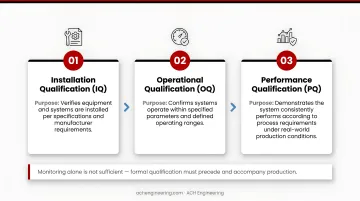
Annual classification testing per ISO 14644-1 is the standard requalification cadence, and cleanroom validation services ensure that documentation is audit-ready, though teams can extend the frequency based on risk assessment and consistent compliance data.
Modular Cleanroom Solutions for ISO 13485-Certified Facilities
Modular cleanroom systems offer an alternative to traditional construction, with significant advantages for medical device manufacturers.
Benefits of Modular Construction
- Faster deployment: Case studies like Project Blackhawk demonstrate that prefabricated cleanrooms can transform existing facilities in significantly less time than stick-built construction. Off-site fabrication allows renovation and construction to occur in parallel.
- Pre-validated components: Factory-manufactured panels, ceiling systems, and HEPA housings arrive with documentation supporting validation efforts.
- Flexibility for expansion: Modular systems can be reconfigured, expanded, or relocated as manufacturing needs evolve—critical for medical device companies adapting to market demands.
- Reduced construction contamination: Manufacturing components off-site minimizes particle generation and contamination risks in existing facilities.
ACH Engineering's Modular Cleanroom Solutions
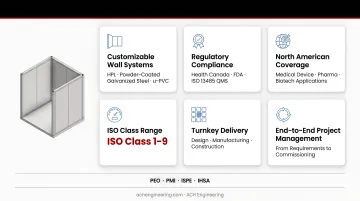
These benefits translate directly into how ACH Engineering structures its modular cleanroom offering. ACH provides turnkey cleanroom solutions designed for medical device, pharmaceutical, and biotech applications across North America, with systems covering ISO Class 1-9 requirements and built to support ISO 13485 quality management systems from day one.
Key features include:
- Customizable wall systems: HPL, powder-coated galvanized steel, and u-PVC options compliant with Health Canada, FDA, and cGMP requirements
- Integrated HVAC: HEPA filtration, controlled pressurization, and environmental controls (18°C-24°C, 30-60% RH)
- Validation support: IQ/OQ/PQ services to ensure regulatory compliance
- Rapid deployment: Design completed in weeks, installation in weeks rather than months
For manufacturers managing evolving product lines, this means cleanroom infrastructure that scales without triggering full revalidation cycles—keeping compliance continuous rather than reactive.
Maintaining Ongoing Compliance: Operations and Quality Management
Once your cleanroom is classified and operational, keeping it compliant is an ongoing commitment — one that spans system maintenance, documentation, and personnel oversight.
Routine Maintenance Requirements
Filter maintenance:
- HEPA filter integrity testing annually or per manufacturer recommendations
- Filter replacement when pressure drop exceeds specifications
- Documentation of all filter changes and testing
System recertification:
- Annual classification testing per ISO 14644-1
- Pressure differential verification
- Airflow pattern studies (as needed)
Cleaning validation:
- Documented cleaning procedures with frequency based on risk assessment
- Environmental monitoring to verify cleaning effectiveness
- Periodic review and revalidation of cleaning protocols
Documentation Requirements
ISO 13485 requires comprehensive documentation integrating cleanroom operations:
Environmental logs:
- Continuous monitoring data for particles, pressure, temperature, humidity
- Microbial monitoring results (for sterile environments)
- Deviation reports and investigations
Beyond environmental logs, two additional documentation streams are mandatory:
- CAPA integration — Environmental deviations feed into the Corrective and Preventive Action system, requiring root cause analysis and effectiveness verification before closure
- Change control — Modifications to cleanroom design, equipment, or procedures must go through documented change control before implementation
Personnel Training Requirements
ISO 13485 Clause 6.2 requires demonstrated competence for personnel whose work affects product quality. For cleanroom operations, this includes:
- Gowning procedures and contamination control practices
- Understanding of environmental monitoring systems
- Recognition of deviations and appropriate responses
- Documented training records with periodic retraining
Competency assessments — not just attendance records — serve as the documented proof auditors look for during ISO 13485 reviews.
Frequently Asked Questions
What is the cleanroom classification for medical device manufacturing?
Medical device cleanrooms typically range from ISO Class 5 to ISO Class 8, with the specific classification determined by device risk class, sterility requirements, and manufacturing process. Class III implantable devices often require ISO 5-7, while Class II devices may operate in ISO 7-8 environments.
Do you need ISO 13485 to manufacture medical devices?
ISO 13485 certification isn't legally required in every jurisdiction, but it demonstrates compliance with quality management requirements under Health Canada's CMDR, the FDA's QMSR, and the EU MDR — and is widely required by customers and regulatory bodies. In the US, the FDA's QMSR now incorporates ISO 13485:2016 by reference, making certification effectively mandatory for North American manufacturers.
What is a manufacturing clean room?
A manufacturing cleanroom is a controlled environment where airborne particle concentration, temperature, humidity, and pressure are regulated to minimize contamination risks during production of sensitive products like medical devices and pharmaceuticals. Classification is based on particle counts per ISO 14644-1 standards.
How often must cleanrooms be recertified?
ISO 14644-2 requires annual classification testing as a baseline, though frequency can be extended based on risk assessment and compliance history. Continuous monitoring of critical parameters — particles and pressure differentials — is required between certifications, with alert and action levels triggering formal investigations.
What pressure differential is required between cleanroom zones?
Health Canada GMP guidance, EU GMP Annex 1, and ISPE all recommend a minimum 10 Pascal pressure differential between adjacent rooms of different cleanliness grades. This positive pressure cascade prevents contamination migration into cleaner areas and requires continuous monitoring.


