
Introduction
Regulatory approval is often the longest and most costliest phase of cleanroom deployment—yet it's consistently underestimated in project planning. For pharmaceutical manufacturers, biotech companies, medical device producers, compounding pharmacies, and lab managers preparing for Health Canada, FDA, ISO 14644, or GMP compliance, the stakes are high.
Approval delays carry a real price tag. Industry analyses estimate that a single day of delay in drug development costs approximately $800,000 in lost sales—a figure that puts even minor timeline slippage in sharp financial focus.
Non-compliance carries even steeper consequences: failed audits, product recalls, facility shutdowns, and loss of manufacturing licenses. These aren't hypothetical risks—FDA warning letters consistently cite procedural failures, inadequate environmental monitoring, and insufficient validation documentation as the primary causes of regulatory action.
This guide covers the full regulatory approval process—from applicable standards and documentation requirements to realistic timelines and how modular cleanroom design can accelerate compliance without compromising the testing and validation regulators require.
Key Takeaways
- Regulatory approval follows four sequential stages: Design Qualification (DQ), Installation Qualification (IQ), Operational Qualification (OQ), and Performance Qualification (PQ)
- North American facilities answer to FDA, ISO (14644 series), EU GMP Annex 1, and Health Canada
- Typical approval timelines range from 9–18 months depending on cleanroom classification, industry sector, and documentation readiness
- Modular cleanrooms cut approval timelines by 60–70% via pre-designed designs, factory acceptance testing, and standardized documentation
- Documentation quality—not hardware—is the primary cause of approval delays
What Is the Regulatory Approval Process for Modular Cleanrooms?
The regulatory approval process is the formal sequence of documentation, testing, and verification required to prove a cleanroom meets applicable standards — FDA, ISO, or GMP — before production can begin. It covers two distinct phases: initial qualification (proving the cleanroom works as designed) and ongoing validation (proving it continues to work over time).
Approval is not a single event. It follows a structured progression through four qualification stages, each building on the last:
- Design Qualification (DQ) — confirms the design meets regulatory and operational requirements
- Installation Qualification (IQ) — verifies the cleanroom was built to specification
- Operational Qualification (OQ) — tests that systems perform correctly under defined conditions
- Performance Qualification (PQ) — confirms consistent performance under actual production conditions
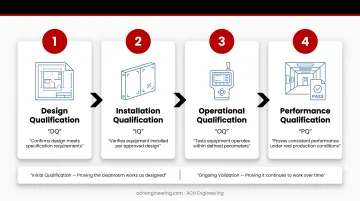
Deficiencies found late often force a return to earlier stages, adding months and budget to the project.
For modular cleanrooms, this process can be accelerated through factory testing and pre-designed designs. But modular construction doesn't earn regulatory exemptions — every cleanroom, regardless of how it's built, must demonstrate compliance through documented evidence before it can be approved for controlled manufacturing.
Why Regulatory Approval Is Critical for Cleanroom Operations
Regulatory approval is legally mandated for industries producing pharmaceuticals, biologics, medical devices, and other products where contamination control directly impacts patient safety. Operating without it puts manufacturing operations at direct risk:
- Regulatory citations and warning letters that can halt production indefinitely
- Product seizures when manufacturing occurs in non-compliant facilities
- Loss of manufacturing licenses that prevent market access
- Failed audits that damage reputation and customer relationships
Approval documentation does more than keep you off a warning letter list — it's your primary defense during inspections. Regulators like Health Canada and the FDA don't pre-approve facilities; they audit them to confirm your records prove compliance with GMP requirements. Strong documentation turns an inspection from a liability into a routine confirmation.
Approval also locks in baseline performance metrics for your cleanroom environment. When particle counts, air pressure differentials, or temperature readings drift from those baselines, you have a documented trigger for investigation — and a clear threshold for when revalidation is required rather than a judgment call.
Key Regulatory Standards and Requirements
FDA Standards (21 CFR Parts 210 and 211)
FDA's Current Good Manufacturing Practice (CGMP) regulations set minimum requirements for pharmaceutical manufacturing facilities, covering environmental controls, documentation systems, and quality management. 21 CFR 211.42 specifically mandates separate or defined areas for aseptic processing, requiring HEPA filtration, positive pressure differentials, and smooth, hard surfaces in critical areas.
The FDA does not "pre-approve" facilities but conducts inspections to verify compliance before granting manufacturing licenses. Their 2004 Aseptic Processing Guidance provides detailed expectations for environmental monitoring, smoke studies to visualize airflow patterns, and validation of critical areas (typically ISO Class 5 environments).
ISO 14644 Series
ISO 14644-1:2015 is the international standard for cleanroom classification based on airborne particle counts. ISO Class 5 through Class 8 are most common for pharmaceutical applications, with Class 5 representing the highest cleanliness level for critical aseptic operations.
ISO 14644-2 covers testing and monitoring requirements to verify cleanroom performance, including recommended pressure differentials of 10-15 Pascals between classified areas. While ISO certification is voluntary, most global buyers and contract manufacturers require it as a baseline for companies serving international markets.
EU GMP Annex 1 (for European Markets)
EU GMP Annex 1 (revised 2022) builds directly on ISO classifications, defining Grade A, B, C, and D environments for sterile manufacturing. Grade A (equivalent to ISO Class 5 "in operation") requires strict unidirectional airflow at 0.36-0.54 m/s velocity, verified through smoke studies.
Companies exporting to Europe must comply with both FDA and EU GMP standards simultaneously. Annex 1 also mandates a comprehensive Contamination Control Strategy (CCS) that ties together facility design, cleaning procedures, gowning protocols, and environmental monitoring into one documented framework.
Health Canada Requirements (for Canadian Facilities)
Health Canada's Good Manufacturing Practices Regulations align closely with FDA standards but include specific documentation and inspection protocols. GUI-0001 and GUI-0119 adopt PIC/S guidance for sterile manufacturing, requiring strict environmental control, pressure differentials, and sanitation programs.
For facilities operating across North American markets, dual compliance with U.S. and Canadian requirements is a practical reality—not an optional extra. ACH Engineering's experience spanning Ontario to Alberta puts that cross-border regulatory coordination directly within their project scope.
Table 1: Regulatory Standards Comparison for Cleanroom Facilities
| Standard | Governing Body | Scope | Classification Basis | Key Requirements | Applies To |
|---|---|---|---|---|---|
| FDA 21 CFR 210/211 | U.S. Food and Drug Administration | Pharmaceutical manufacturing in the U.S. | GMP compliance (Grades A-D) | Aseptic processing controls, HEPA filtration, positive pressure differentials, inspection-based approval | U.S. pharmaceutical and biotech manufacturers |
| ISO 14644 | International Organization for Standardization | International cleanroom classification | Airborne particle count (ISO Class 1-9) | Particle count testing, pressure differentials (10-15 Pa), periodic requalification | Any industry requiring controlled environments globally |
| EU GMP Annex 1 | European Medicines Agency (EMA) | Sterile pharmaceutical manufacturing for EU markets | ISO-aligned Grades A-D | Unidirectional airflow (0.36-0.54 m/s), Contamination Control Strategy (CCS), smoke studies | EU market pharmaceutical and biotech producers |
| Health Canada GMP | Health Canada / PIC/S | Canadian pharmaceutical manufacturing | Aligned with FDA and PIC/S | GUI-0001 and GUI-0119 compliance, pressure differentials, sanitation programs | Canadian pharmaceutical manufacturers and dual-market exporters |
How the Regulatory Approval Process Works
The approval process moves sequentially from design documentation through installation verification, operational testing, and performance validation under actual production conditions. Each stage builds on the previous one, creating a documented trail of evidence that proves compliance.
Design Qualification (DQ)
DQ involves documenting that the cleanroom design meets user requirements and applicable standards before construction begins. This is where regulatory compliance is embedded into the project, not added after the fact.
Key DQ deliverables include:
- User Requirement Specifications (URS) defining functional needs
- Design specifications showing how requirements will be met
- Equipment specifications with regulatory compliance features
- Risk assessments identifying potential contamination sources
- Traceability matrices linking requirements to design elements
Installation Qualification (IQ)
IQ verifies that all cleanroom components—walls, ceilings, HVAC systems, HEPA filters, monitoring equipment—are installed correctly according to approved designs. This stage documents the "as-built" condition of the facility.
IQ documentation includes:
- Equipment serial numbers and model specifications
- Calibration certificates for monitoring instruments
- Installation checklists verifying proper assembly
- Material certificates confirming component specifications
- Utility connection verification (electrical, HVAC, controls)
Operational Qualification (OQ)
OQ tests cleanroom systems under controlled conditions (at-rest state with no personnel or production activities) to verify they meet design specifications. This stage proves the systems work as intended when isolated from operational variables.
OQ testing covers five core parameters:
- Airflow velocity and volume measurements to verify design specifications
- HEPA filter integrity testing (≤0.01% penetration per FDA guidance)
- Pressure differential verification (typically 10–15 Pascals between zones)
- Temperature and humidity mapping across the entire cleanroom
- Particle count testing to verify ISO classification at-rest

Performance Qualification (PQ)
PQ tests the cleanroom under actual operating conditions with personnel, equipment, and simulated production activities. This stage confirms the cleanroom maintains specifications during normal use and can consistently support compliant manufacturing.
PQ activities include:
- Environmental monitoring under dynamic conditions
- Smoke studies demonstrating airflow patterns during interventions
- Media fills or aseptic process simulations
- Recovery time testing (how quickly the room returns to specification after disturbances)
- Worst-case scenario testing with maximum personnel and equipment loads
ACH Engineering's turnkey cleanroom model covers each of these stages — from early design documentation through installation protocols and qualification support — so compliance is built into the project schedule rather than addressed as an afterthought. This reduces the risk of discovering gaps late in the process, when they're most costly to address.
Key Factors That Affect the Regulatory Approval Process
Cleanroom Classification Level
Higher-grade cleanrooms (ISO Class 5, FDA Grade A/B) require significantly more rigorous testing, documentation, and validation than lower classifications. Sterile manufacturing facilities face the most stringent approval requirements, including extensive smoke studies to visualize unidirectional airflow and demonstrate sweeping action away from products.
ISO Class 8 environments with turbulent airflow are faster to validate because they don't require the detailed airflow visualization studies mandated for unidirectional flow zones. The difference can add 2-3 months to validation timelines for ISO Class 5 facilities.
Industry Sector and Product Type
Pharmaceutical facilities producing sterile injectables face different requirements than medical device manufacturers or compounding pharmacies. Biologics and cell/gene therapy facilities often require additional biocontainment considerations and specialized environmental monitoring programs.
Each sector has specific regulatory expectations. Compounding pharmacies must meet USP <797>/<800> standards, while medical device manufacturers follow different FDA guidance than pharmaceutical producers. Mapping your sector's requirements before design begins is what keeps qualification on schedule and avoids expensive mid-project corrections.
Documentation Readiness
Incomplete or poorly organized documentation is the leading cause of approval delays. Analysis of FDA Form 483 observations reveals that procedural and documentation failures—such as inadequate investigations, missing written procedures, and insufficient validation documentation—frequently outnumber technical hardware failures.
Critical documentation includes:
- Validation Master Plans (VMP) outlining the overall validation strategy
- Standard Operating Procedures (SOPs) for all critical operations
- Test protocols with acceptance criteria defined before testing
- Calibration records for all monitoring equipment
- Training records demonstrating personnel competency

Modular vs. Stick-Built Construction
Modular cleanrooms can accelerate approval through Factory Acceptance Testing (FAT) conducted before site delivery. FAT can verify up to 30% of system functionality off-site in controlled conditions, identifying defects when they're easiest and cheapest to fix.
Pre-designed modular designs often come with standardized documentation packages that simplify the approval process. These templates have been proven through previous installations, reducing the documentation development time and providing regulators with familiar, proven formats.
Regulatory Authority Coordination
Early engagement with Health Canada reviewers, FDA inspectors, or other relevant authorities clarifies expectations and prevents surprises during final inspections. Both Health Canada and the FDA encourage manufacturers to align facility design and quality systems well before the inspection stage — a step that consistently reduces last-minute findings.
ACH Engineering supports this coordination process through direct experience with Canadian and US compliance requirements. Active membership in ISPE keeps the team current with evolving pharmaceutical engineering standards, which translates into guidance that's grounded in what regulators actually expect to see.
Common Issues and How to Avoid Them
Inadequate Design Documentation
Many projects fail initial reviews because design specifications don't clearly link user requirements to regulatory standards. Reviewers can't verify compliance if the documentation doesn't explicitly show how each design element satisfies specific regulatory requirements.
To avoid this:
- Hold design review meetings with quality assurance and regulatory teams before finalizing plans
- Create traceability matrices that map each user requirement to design specifications and applicable regulatory standards
Rushed Installation and Testing
Compressed timelines lead to incomplete IQ/OQ testing and documentation gaps that delay approval. Skipping verification steps or conducting tests without approved protocols creates compliance gaps that must be addressed before approval.
To avoid this:
- Build realistic timelines that account for equipment lead times, testing schedules, and documentation review cycles
- Budget extra time for unexpected issues, which are virtually guaranteed in complex builds
- Plan for sequential testing rather than compressing parallel activities
Lack of Cross-Functional Coordination
Approval requires input from engineering, quality assurance, operations, and regulatory affairs. Siloed planning causes misalignment between what engineering builds and what quality assurance can validate.
To avoid this:
- Establish a project steering committee in the design phase with representatives from all stakeholder groups
- Hold regular coordination meetings throughout the project to maintain alignment on requirements, timelines, and acceptance criteria
When to Start the Regulatory Approval Process
Regulatory planning should begin during the conceptual design phase, not after construction is complete. Retrofitting compliance into a finished facility can cost three to five times more than building it in from day one — and delays regulatory approval by months.
Engage regulatory consultants or experienced cleanroom providers during User Requirement Specification (URS) development. This ensures compliance requirements are woven into the design before a single panel is ordered — not reconciled after the fact.
General timeline expectations:
- Design and documentation development: 6–12 months
- Installation and qualification testing: 3–6 months
- Regulatory review and inspection scheduling: 2–4 months
Total timeline: 9–18 months from design start to approval.
For projects with tighter deadlines, modular construction compresses that timeline by 60–70% through three structural advantages:
- Parallel processing: Modules are built off-site while site preparation runs simultaneously
- Faster installation: 2–4 weeks on-site vs. 3–6 months for traditional builds
- Streamlined validation: Factory Acceptance Testing (FAT) and pre-validated designs reduce qualification cycles
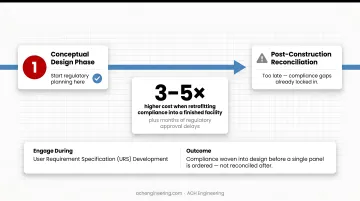
ACH Engineering builds regulatory requirements into the URS stage for exactly this reason — so cleanroom validation documentation is ready when installation finishes, not months after.
Conclusion
The regulatory approval process is a structured sequence of design qualification, installation qualification, operational qualification, and performance qualification that verifies cleanroom compliance with Health Canada, FDA, ISO, and GMP standards. While time-intensive, proper planning and documentation prevent costly delays and failed inspections that can halt production for months.
Modular cleanroom solutions accelerate approval through factory testing, pre-designed designs with documented compliance histories, and standardized documentation packages that cut development time. These advantages can reduce time-to-market by 60-70% while maintaining full regulatory compliance — without shortcutting the qualification steps that regulators require.
Those gains only hold when documentation receives the same rigor as physical construction — it is, after all, the primary deliverable regulators inspect. Embedding compliance into initial designs, maintaining cross-functional coordination throughout the project, and working with providers who understand Canadian and North American regulatory requirements allows manufacturers to achieve faster approval without compromising the validation standards that protect product quality and patient safety.
Frequently Asked Questions
How long does the regulatory approval process take for a modular cleanroom?
Typically 9-18 months from design start to final approval, though modular construction can reduce this by 3-6 months compared to stick-built facilities. Factory acceptance testing identifies issues early, installation runs 2-4 weeks versus 3-6 months for traditional builds, and pre-designed designs come with proven compliance documentation already in place.
What are the most common reasons for regulatory approval delays?
The top causes are incomplete documentation packages, failed qualification tests requiring system modifications, inadequate training records, and poor coordination between engineering and quality assurance teams. Documentation failures outnumber hardware failures in FDA inspection observations.
Do I need separate approvals for FDA and ISO certification?
ISO 14644 certification is voluntary but demonstrates compliance with international standards. FDA compliance is mandatory for pharmaceutical manufacturing in the U.S., while Health Canada governs equivalent requirements for Canadian facilities. Many manufacturers pursue both ISO certification and the applicable regulatory approval to satisfy global market requirements and meet international best practices.
Can a modular cleanroom be pre-certified before installation?
Factory acceptance testing (FAT) verifies system performance before delivery and can cover up to 30% of system functionality. Final regulatory approval still requires site acceptance testing (SAT) and performance qualification (PQ) at the installed location. Transportation can affect filter seals, and site-specific conditions must be independently verified.
What documentation is required for regulatory approval?
A complete regulatory submission typically requires:
- Validation Master Plan (VMP)
- User Requirement Specifications (URS)
- Design Qualification (DQ) report
- Installation Qualification (IQ) protocols and reports
- Operational Qualification (OQ) protocols and reports
- Performance Qualification (PQ) protocols and reports
- Standard Operating Procedures (SOPs) for all critical operations
How often do cleanrooms need to be revalidated after initial approval?
EU GMP Annex 1 mandates revalidation every 6 months for Grade A/B (ISO Class 5) and 12 months for Grade C/D (ISO Class 7/8). Revalidation is also triggered by significant modifications to facilities, equipment, or processes — and ongoing environmental monitoring can flag the need earlier if performance trends drift from established baselines.


