
Introduction: Why ISO 14644 and GMP Cleanroom Standards Matter
Cleanroom standards aren't optional guidelines—they're regulatory requirements that protect product quality, patient safety, and manufacturing consistency. When facilities fail to meet these standards, the consequences are immediate and severe.
Recent FDA enforcement data reveals the business impact. Bio-Medical Pharmaceutical Manufacturing Corporation received a warning letter in February 2026 for lacking classified cleanrooms and ISO 5 conditions for aseptic filling, resulting in voluntary recalls. Empower Pharmacy faced citations in April 2025 for inadequate environmental monitoring in critical ISO 5 areas.
A single contaminated batch in sterile manufacturing costs between $1-2 million, not counting production shutdowns, failed audits, and reputational damage.
This guide addresses both ISO 14644 (international air cleanliness classification) and GMP Annex 1 (pharmaceutical regulatory requirements). You'll learn:
- How these standards relate and where they differ
- What changed in recent revisions and why it matters
- Practical steps to achieve and maintain compliance
TLDR: Key Takeaways
- ISO 14644 classifies air cleanliness (ISO 1-9) by particle count; pharma uses ISO 5-8
- GMP grades (A-D) add operational requirements: microbial limits and Contamination Control Strategy
- 2015 ISO revision changed sampling methods and removed 5μm limits from ISO Class 5
- Successful compliance demands integrated HVAC design, validated materials, and continuous monitoring
- Document everything: qualification protocols are essential for regulatory approval
Understanding ISO 14644 Cleanroom Classification
What ISO 14644 Standards Cover
ISO 14644 is a multi-part international standard series that governs cleanroom classification, monitoring, testing, design, construction, and operations.
Part 1 (ISO 14644-1:2015) focuses on classification by airborne particle concentration, while Part 2 addresses monitoring requirements for continued compliance.
The standard defines three occupancy states:
- As-built: Empty facility with services connected and functioning
- At-rest: Equipment installed and operating, no personnel present
- Operational: Normal working conditions with staff and active processes
ISO Cleanroom Classes Explained
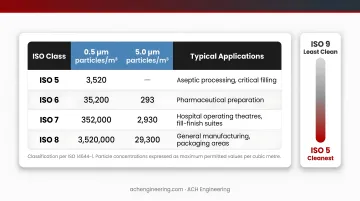
ISO classes range from ISO 1 (cleanest) to ISO 9 (least clean), with classification based on maximum permitted particle concentrations per cubic metre at different particle sizes.
ISO Classification Table (Selected Classes)
| ISO Class | 0.5 µm particles/m³ | 5.0 µm particles/m³ | Typical Applications |
|---|---|---|---|
| ISO 5 | 3,520 | (M descriptor) | Aseptic processing, critical filling |
| ISO 6 | 35,200 | 293 | Pharmaceutical preparation |
| ISO 7 | 352,000 | 2,930 | Less critical preparation, packaging |
| ISO 8 | 3,520,000 | 29,300 | Component handling, warehousing |
Most pharmaceutical applications use ISO Classes 5-8, with ISO 5 reserved for critical aseptic processing areas where sterile products contact open containers or critical surfaces.
Key Changes in ISO 14644-1:2015 Revision
The 2015 revision introduced four significant changes that impact pharmaceutical compliance:
Sampling Point Calculation: The formula-based approach was replaced with a table-based lookup system (Annex A), typically increasing required sampling locations for most cleanrooms.
This change simplifies calculations but demands more comprehensive testing.
UCL Elimination: The 95% Upper Confidence Limit calculation requirement was removed. Each sampling point must now independently meet classification limits—no statistical averaging across locations.
5μm Particle Limits: The 5μm particle limit was removed from the ISO Class 5 classification table.
However, because GMP Annex 1 still requires 5μm monitoring for Grade A/B areas, the "M descriptor" notation (ISO M) was introduced to track macroparticles when regulatory agencies mandate it.
Equipment Requirements: Particle counters must comply with ISO 21501-4 calibration standards, and sampling tube length is limited to 1 metre maximum to minimize particle loss.
Classification Testing Requirements
Core classification tests include:
- Airborne particle counting at specified locations using calibrated particle counters
- Airflow velocity and uniformity measurements to verify air change rates
- HEPA filter integrity testing (leak testing) to confirm filtration performance
- Pressure differential verification between adjacent zones
Sampling locations are determined using the revised table-based approach from ISO 14644-1:2015 Annex A. Minimum sample volume requirements depend on cleanroom class and particle size—ISO 5 cleanrooms require larger sample volumes than ISO 7 or 8 to achieve statistical confidence at lower particle concentrations.

Ongoing Monitoring and Re-classification
Cleanrooms must be re-classified periodically, typically every 6-24 months depending on risk assessment and regulatory requirements.
According to ISO 14644-2:2015, annual re-classification is standard, though this frequency may be extended based on documented risk assessment and consistent monitoring data.
Classification vs. Routine Monitoring:
- Classification testing: Comprehensive, periodic verification using the full sampling plan
- Routine monitoring: Continuous or frequent sampling at targeted high-risk locations
Establish documented monitoring programmes with defined alert and action limits, investigation procedures when limits are exceeded, and trending analysis to detect deterioration before classification failures occur.
GMP Annex 1 Requirements for Pharmaceutical Cleanrooms
Understanding GMP and Annex 1
Good Manufacturing Practice (GMP) is the regulatory framework ensuring medicinal products are consistently produced and controlled to quality standards. EU GMP Annex 1, revised in 2022 and effective August 2023, specifically addresses sterile medicinal product manufacture.
Regulatory authorities including MHRA and FDA enforce these standards across pharmaceutical manufacturing operations.
The revised Annex 1 mandates a documented Contamination Control Strategy (CCS)—a comprehensive quality system integrating facility design, personnel practices, cleaning, utilities, and monitoring.
This shifts compliance from checklist exercises to scientific, risk-based strategies demonstrating continuous control.
GMP Cleanroom Grades A, B, C, and D
These grades align cleanroom environments with the criticality of pharmaceutical operations performed within them:
- Grade A: Critical aseptic operations (filling, making sterile connections)
- Grade B: Background environment for Grade A zones
- Grade C: Less critical preparation steps
- Grade D: Component handling and washing
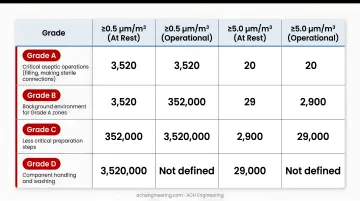
GMP Particle Limits for Monitoring
| Grade | ≥0.5 µm/m³ (At Rest) | ≥0.5 µm/m³ (Operational) | ≥5.0 µm/m³ (At Rest) | ≥5.0 µm/m³ (Operational) |
|---|---|---|---|---|
| A | 3,520 | 3,520 | 29 | 29 |
| B | 3,520 | 352,000 | 29 | 2,930 |
| C | 352,000 | 3,520,000 | 2,930 | 29,300 |
| D | 3,520,000 | Not predetermined | 29,300 | Not predetermined |
Microbiological Limits (Monitoring)
| Grade | Air Sample (CFU/m³) | Settle Plates (CFU/4 hours) | Contact Plates (CFU/plate) | Glove Print (CFU/glove) |
|---|---|---|---|---|
| A | <1 | <1 | <1 | <1 |
| B | 10 | 5 | 5 | 5 |
| C | 100 | 50 | 25 | - |
| D | 200 | 100 | 50 | - |
Unlike ISO standards, GMP grades include microbiological limits through air sampling, surface contact plates, and personnel monitoring.
Contamination Control Strategy Requirements
The revised Annex 1 mandates a written CCS documenting all measures to prevent contamination throughout the product lifecycle.
Key CCS Elements:
- Design rationale for facilities and equipment based on contamination risk assessment
- Qualification protocols for air handling systems and their ongoing validation
- Validated cleaning and disinfection programs with documented effectiveness
- Training programs covering gowning qualification and aseptic technique
- Strategies for material flow control and operational segregation
Risk assessments (FMEA, HACCP-style approaches) must identify contamination risks and justify control measures. The CCS isn't a static document—it requires ongoing review and updates based on monitoring trends and operational changes.
Operational and Procedural Requirements
Personnel and Process Controls
Beyond air quality specifications, GMP mandates rigorous personnel and process controls:
- Personnel gowning qualification with documented competency assessment
- Aseptic technique validation through regular observation and requalification
- Media fills (process simulation tests) demonstrating sterility assurance
- Cleaning validation proving effective removal of residues and microorganisms
- Environmental monitoring programmes with continuous data collection
Airflow and Environmental Protection
The critical concept of "first air" protection ensures HEPA-filtered air reaches product and critical surfaces before contacting any potential contamination source. Airflow visualization studies (smoke studies) must demonstrate proper air patterns.
Documentation and Data Integrity
Documentation requirements include batch records, deviation investigations, change control, and annual product quality reviews. Regulatory inspection readiness demands data integrity compliance following ALCOA+ principles: Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, and Available.
Key Updates in Revised Annex 1 (2022)
The 2022 revision emphasizes:
- Quality Risk Management (QRM) integrated throughout contamination control decisions
- Continuous monitoring explicitly required for Grade A zones during all critical operations (including setup); strongly recommended for Grade B
- Airflow visualization studies to demonstrate first air protection and proper air patterns
- Enhanced data integrity expectations with real-time data acquisition and trending
ISO vs GMP: Understanding the Relationship
ISO 14644 and GMP Annex 1 serve different but complementary purposes in pharmaceutical cleanroom operations. ISO 14644 is a technical standard focused on measuring and classifying air cleanliness, while GMP provides a comprehensive regulatory framework governing all aspects of pharmaceutical manufacturing quality, including documentation, personnel training, and operational controls.
Understanding how these classifications align helps facilities plan their cleanroom designs and operational protocols:
| GMP Grade | ISO Classification (At Rest) | ISO Classification (Operational) |
|---|---|---|
| Grade A | ISO 5 | ISO 5 |
| Grade B | ISO 5 | ISO 7 |
| Grade C | ISO 7-8 | ISO 7-8 |
| Grade D | ISO 8 | ISO 8 |

Critical distinction: These are approximations. GMP limits always take regulatory precedence when they differ from ISO classifications.
Pharmaceutical cleanrooms must meet both standards simultaneously:
- ISO 14644 defines classification methodology and testing procedures
- GMP Annex 1 establishes operational controls and compliance expectations
Designing and Building Compliant Cleanroom Facilities
Facility Layout and Design Principles
Pressure Cascade Design: Maintain 5-15 Pa positive pressure differentials from lower to higher cleanliness grades.
ISPE guidance recommends minimum 10 Pa between different grades and at least 5 Pa between rooms of the same classification to prevent contamination migration.
Segregation Principles:
- Separate personnel and material flows with dedicated corridors
- Airlocks with interlocked doors between grade transitions
- Dedicated gowning areas appropriately sized for the number of personnel
- Waste removal routes that minimize cross-contamination risk
Effective space planning supports equipment placement, maintenance access, material staging, and personnel movement. This layout approach maintains proper air patterns while avoiding dead zones where air stagnates.
Cleanroom Envelope and Construction Materials
Surface Requirements:
- Smooth, non-shedding, non-porous, cleanable surfaces
- Flush joints with minimal gaps or ledges
- Coved corners eliminating sharp 90-degree angles
- Minimal particle traps or horizontal surfaces
Materials must be compatible with cleaning agents, disinfectants, and fumigation processes (hydrogen peroxide vapour, formaldehyde).
Options include modular panel systems, epoxy-coated walls, sealed concrete floors with epoxy coatings, and suspended ceiling systems designed for cleanroom use. Modular systems like those offered by ACH Engineering provide GMP-compliant components with seamless coving that eliminates sharp corners in compliance with cGMP requirements, meeting both ISO and GMP standards.
HVAC System Design and Requirements
Critical system components ensure proper air quality and environmental control throughout the facility.
Core HVAC Components:
- HEPA or ULPA filters (minimum H13/H14 for pharmaceutical use, ≥99.95% efficiency)
- Air handling units with precise temperature and humidity controls
- Ductwork designed to minimize particle accumulation
Air Change Rates:
- Grade C/D (ISO 7-8): Typically >20 air changes per hour
- Grade A/B (ISO 5): Unidirectional (laminar) flow at 0.36-0.54 m/s velocity rather than ACH
Environmental Controls:
- Temperature: Typically 18-22°C
- Humidity: 35-55% RH
- Impact: Product stability, microbial growth control, and personnel comfort in full cleanroom gowning
Doors, Windows, and Penetrations
Cleanroom Doors:
- Flush-mounted with no protruding hardware
- Self-closing mechanisms
- Interlocked where required between grade transitions
- Proper seals to maintain pressure differentials
Vision Panels:
- Flush-mounted for easy cleaning
- Tempered glass or polycarbonate materials that don't shed particles
- Minimal frames or ledges
Utility Penetrations:
- Sealed penetrations for electrical, plumbing, and process piping
- Minimal protrusions into cleanroom space
- Accessible for maintenance without compromising cleanroom integrity
Material Selection and Documentation
All materials require documented cleanroom compatibility, including certificates of compliance and extractables/leachables data for critical applications.
Maintain material traceability and change control to ensure validated materials aren't substituted during construction. Document all construction materials and methods in Design Qualification (DQ) and Installation Qualification (IQ) protocols for regulatory inspection readiness.
Environmental Monitoring and Testing Requirements
Maintaining ISO 14644 and GMP compliance requires continuous environmental monitoring to verify your cleanroom performs within specified parameters. This monitoring validates that contamination control measures remain effective during operations.
Environmental monitoring falls into two categories:
Non-viable Monitoring (Particle Counting)
- Continuous or at defined intervals using calibrated particle counters
- Sampling tube length maximum 1 metre
- Systems log data with timestamps and location identifiers for full traceability
Viable Monitoring (Microbial Sampling)
- Active air samplers collecting microorganisms on growth media
- Settle plates exposing media to ambient air for defined periods
- Surface contact plates sampling work surfaces and equipment
- Personnel monitoring via glove prints and gown contact plates
Monitoring Frequencies by Cleanroom Grade
Monitoring frequency depends on your cleanroom classification and the criticality of operations:
- Grade A critical areas: Continuous particle monitoring during operations
- Grade B: Continuous or daily monitoring recommended
- Grade C/D: Weekly to monthly based on risk assessment
- Viable monitoring: Daily to weekly depending on grade and risk
Data Management and Response Protocols
Effective monitoring requires more than just collecting data:
- Establish alert and action limits based on baseline qualification data from your initial cleanroom validation
- Investigate all excursions immediately and document root cause analysis
- Trend results over time to identify patterns before they become compliance issues
- Document all monitoring activities and corrective actions in compliance with ALCOA+ data integrity principles (Attributable, Legible, Contemporaneous, Original, Accurate, plus Complete, Consistent, Enduring, Available)
Many pharmaceutical and biotech facilities in North America integrate automated monitoring systems that provide real-time alerts when parameters drift toward action limits, enabling proactive intervention before contamination events occur.
Validation and Qualification Process
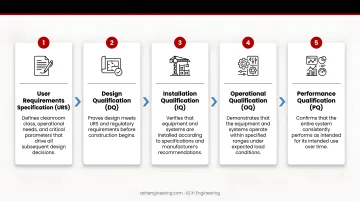
Cleanroom validation follows a structured lifecycle that proves your facility meets ISO 14644 and GMP requirements from initial design through daily operations. Each stage builds on the previous one, creating a documented trail of compliance.
Lifecycle Stages:
- User Requirements Specification (URS): Defines cleanroom class, operational needs, and critical parameters that drive all subsequent design decisions
- Design Qualification (DQ): Proves design meets URS and regulatory requirements through engineering calculations and risk assessments
- Installation Qualification (IQ): Verifies correct installation, materials, and documentation—inspectors check that equipment matches specifications and is properly calibrated
- Operational Qualification (OQ): Confirms performance at rest conditions without personnel, testing parameters like airflow velocity, pressure differentials, and filter integrity
- Performance Qualification (PQ): Verifies control during normal operations with personnel and processes active, simulating worst-case production scenarios
ACH Engineering's project management services guide facilities through the entire validation lifecycle, from URS development to final PQ documentation, helping ensure smooth regulatory inspections.
Ongoing Verification Requirements
Initial qualification is just the beginning. Maintaining compliance requires regular requalification and continuous monitoring to prove your cleanroom remains within specification.
Requalification Schedule:
- Every 6 months for Grade A/B areas
- Annually for Grade C/D areas
- After significant changes (HVAC modifications, layout changes, process updates)
Continuous Compliance:
- Real-time monitoring with automated data trending
- Annual review of environmental data and contamination control strategy effectiveness
- Documentation updates reflecting any facility or process changes
Common Compliance Challenges and Solutions
Frequent Pitfalls
Confusing ISO classes with GMP grades creates serious compliance gaps. Specifying "ISO 5" when you need "Grade A" misses microbiological limits and operational requirements.
Always specify the GMP grade for pharmaceutical applications, with ISO classification as supporting documentation.
Inadequate detailing fails inspections. Overlooking flush detailing and coved corners creates particle traps that auditors flag immediately. Specify seamless wall-ceiling and wall-floor connections from the design phase.
Documentation gaps delay validation and create inspection findings. Incomplete records of design decisions and material selections become major obstacles during regulatory review. Maintain comprehensive documentation packages from URS through PQ.
Challenges with 2015 ISO Revision
The 2015 revision introduced changes that catch many facilities off guard:
- The table-based approach typically requires more sampling points, potentially revealing contamination issues previously undetected—budget additional testing time and resources
- Elimination of UCL calculation means all points must pass independently with no statistical averaging—address marginal locations early rather than relying on calculations
- The 5μm "M descriptor" notation requires updated SOPs and training—document your approach to 5μm monitoring in your CCS to satisfy both ISO and GMP requirements
Solutions
- Work with experienced cleanroom designers during initial project planning—not after layouts are finalized
- Conduct FMEA risk assessments before locking in design decisions
- Implement change control from day one of construction to track all modifications
- Build comprehensive documentation packages as you go, from URS through PQ
- Establish monitoring programs with clear response procedures for excursions
Frequently Asked Questions
What is a GMP cleanroom?
A GMP cleanroom is a controlled environment for pharmaceutical manufacturing that meets Good Manufacturing Practice regulatory requirements. It uses specific air cleanliness grades (A, B, C, D), environmental monitoring programs, and contamination control procedures to ensure product quality.
What is the ISO 14644 series of standards?
ISO 14644 is an international standard series for classifying, testing, and operating cleanrooms based on airborne particle concentration. Part 1 covers classification methodology, while Part 2 addresses ongoing monitoring requirements.
How are GMP cleanrooms classified?
GMP cleanrooms use a grade system (A, B, C, D) based on maximum permitted airborne particles at 0.5μm and 5.0μm sizes in both "at rest" and "in operation" states. Each grade includes microbiological limits for viable particles and specific operational controls required by regulatory authorities.
What are the GMP requirements for a cleanroom?
Key GMP requirements include:
- Appropriate grade classification for the manufacturing process
- Documented Contamination Control Strategy
- Validated HVAC systems with proper air quality and pressure differentials
- Environmental monitoring programs with continuous data collection
- Personnel training, gowning procedures, and cleaning validation
- Comprehensive documentation meeting ALCOA+ data integrity principles
What's the difference between ISO 14644 and GMP Annex 1?
ISO 14644 provides the technical methodology for classifying and testing air cleanliness, while GMP Annex 1 establishes regulatory requirements for pharmaceutical manufacturing including operational controls, microbiological limits, and documentation. Pharmaceutical facilities must comply with both—ISO for classification procedures and GMP for operational standards.
How often must cleanrooms be re-classified?
Re-classification frequency depends on cleanroom grade:
- Grade A and B: Every 6 months maximum
- Grade C and D: Annually
- Also required after HVAC changes, layout modifications, or when monitoring trends indicate issues


