
Introduction: The Critical Role of Quality Assurance in Cleanroom Operations
A single cleanroom QA failure can shut down a production line, trigger a regulatory investigation, or—in the worst cases—harm patients. Cleanroom quality assurance is the systematic framework that keeps those risks in check, ensuring controlled environments meet regulatory standards and protect product integrity across pharmaceutical, biotech, and medical device operations.
Recent FDA enforcement data confirms that environmental monitoring and facility design remain top citation areas in warning letters issued during 2024 and 2025. Violations consistently point to inadequate materials of construction, poor facility maintenance, and insufficient monitoring frequencies.
The financial stakes are real. Contaminated sterile batches can cost $1–2 million USD or more, and regulatory sanctions can halt production entirely until remediation plans are approved and implemented.
The 2012 NECC fungal meningitis outbreak — resulting in over 60 deaths — illustrates how cleanroom QA failures can move from a regulatory problem to a patient safety crisis. This guide covers how to build a QA program that prevents both.
TLDR:
- QA failures trigger recalls, regulatory sanctions, and costs exceeding $1M USD per incident
- FDA warning letters in 2024–2025 most often cite environmental monitoring and facility design gaps
- Effective QA integrates preventive controls from initial design through ongoing operations
- Key standards include ISO 14644, FDA regulations, EU GMP Annex 1, and USP requirements
- Full implementation typically takes 3–6 months with proper planning and resources
Understanding Cleanroom Quality Assurance: Foundations and Principles
Defining Cleanroom QA
Cleanroom quality assurance covers the policies, procedures, and practices that ensure controlled environments consistently meet cleanliness classifications and regulatory requirements. Traditional manufacturing QA focuses on product quality alone — cleanroom QA must address environmental integrity at the same time. Facility performance directly impacts product safety, making this dual focus non-negotiable.
QA vs. QC: Critical Distinctions
Quality Assurance (QA) represents the proactive, system-focused approach:
- Designs contamination control strategies before operations begin
- Establishes facility qualification protocols (DQ/IQ/OQ/PQ)
- Develops comprehensive training programs
- Creates preventive maintenance schedules
- Implements validation procedures for sterilization processes
Quality Control (QC) focuses on detective, product-level verification:
- Performs daily environmental monitoring
- Conducts particle counting and microbial sampling
- Tests product bioburden levels
- Verifies batch records for accuracy
- Investigates out-of-specification results and deviations
21 CFR 211.22 mandates a Quality Control Unit with authority to approve or reject procedures and products, ensuring "no errors have occurred or, if errors have occurred, that they have been fully investigated."
That regulatory mandate reflects a broader shift in how QA frameworks are designed — one increasingly driven by risk rather than fixed rules.
Risk-Based Approach to Cleanroom QA
Regulators have moved away from one-size-fits-all requirements toward risk-proportionate QA frameworks. The 2023 ICH Q9(R1) revision reinforces quality risk management as a core principle across pharmaceutical quality systems. Separately, the 2022 EU GMP Annex 1 requires formal Contamination Control Strategies (CCS) that "define all critical control points and assess the effectiveness of all controls."
Risk assessment determines:
- Environmental monitoring frequencies and locations
- Alert and action limit thresholds
- Requalification intervals
- Personnel gowning requirements
- Cleaning validation scope
These risk-driven decisions feed directly into how a QA program is structured — and what it's ultimately trying to protect against.
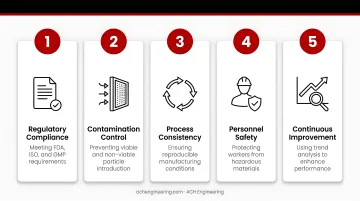
Core QA Program Objectives
Effective cleanroom QA programs pursue five interconnected objectives:
- Regulatory Compliance - Meeting FDA, ISO, and GMP requirements
- Contamination Control - Preventing viable and non-viable particle introduction
- Process Consistency - Ensuring reproducible manufacturing conditions
- Personnel Safety - Protecting workers from hazardous materials
- Continuous Improvement - Using trend analysis to enhance performance
Key Stakeholders
Successful QA programs require collaboration across multiple functions:
- Quality Assurance Managers (program oversight and regulatory liaison)
- Facility Engineers (HVAC systems and environmental controls)
- Production Personnel (daily operations and deviation reporting)
- Regulatory Affairs (compliance strategy and audit preparation)
- External Auditors (third-party verification and certification)
Key Compliance Standards and Regulations
Key Compliance Standards and Regulations
ISO 14644 Series Standards
Two core standards govern global cleanroom classification and monitoring:
- ISO 14644-1:2015 — Defines air cleanliness classifications by particle concentration, specifying maximum allowable particles per cubic meter for sizes ≥0.1 μm to ≥5 μm; each sampling location evaluated at 95% confidence
- ISO 14644-2:2015 — Establishes risk-based monitoring plans (rather than fixed schedules) to prove continued compliance through continuous control verification
FDA Regulations for Pharmaceuticals
21 CFR Parts 210 and 211 govern pharmaceutical manufacturing with specific cleanroom requirements:
- Facility Design (§211.42) — Aseptic processing areas must have smooth cleanable surfaces, temperature/humidity controls, and HEPA-filtered air under positive pressure
- Environmental Monitoring (§211.42(c)(10)(iv)) — Requires systems that track conditions in aseptic processing zones throughout production
- Microbiological Control (§211.113) — Written procedures must prevent contamination; sterilization processes require formal validation
FDA guidance recommends positive pressure differentials of 10-15 Pascals (0.04-0.06 inches water column) between cleanrooms of different classifications and at least 20 air changes per hour for ISO 8 supporting rooms.
FDA 21 CFR Part 820 for Medical Devices
Part 820 applies when environmental conditions could directly affect device quality. Key requirements include:
- Establish written procedures controlling environmental conditions throughout manufacturing
- Periodically inspect environmental control systems to confirm continued performance
- Validate any process that cannot be fully verified after the fact — sterilization being the primary example
EU GMP Annex 1 Requirements
The 2022 revision introduced mandatory Contamination Control Strategies and continuous monitoring for critical zones:
Grade Classification System:
- Grade A — Aseptic filling and other high-risk operations; continuous particle monitoring required during production; ≤3,520 particles/m³ (≥0.5 μm)
- Grade B — Immediate background to Grade A zones; monitoring frequency determined by risk assessment
- Grades C & D — Supporting clean areas for progressively less critical manufacturing steps
The revision emphasizes monitoring "in operation throughout all critical stages of processing" rather than only at-rest conditions.
USP <797> and <800> Standards
USP <797> governs sterile compounding, requiring ISO Class 5 Primary Engineering Controls (PEC) located within ISO Class 7 buffer rooms.
USP <800> addresses hazardous drug handling with specific pressure requirements:
- Buffer rooms: ISO Class 7, negative pressure of 0.01-0.03 inches water column relative to adjacent spaces
- Anterooms: ISO Class 7, positive pressure of at least 0.02 inches water column to adjacent spaces
Industry-Specific Standards
Beyond pharmaceutical and medical applications, industry-specific standards address additional control requirements:
- SEMI Standards — Semiconductor cleanrooms must address electrostatic discharge control and ultrapure water systems in addition to particle limits
- IEST-RP-CC034.5 — Covers HEPA/ULPA filter leak testing procedures
- IEST-RP-CC006.3 — Defines cleanroom performance characterization methods
Essential Components of a Cleanroom QA Program
Environmental Monitoring Program
Effective environmental monitoring anchors cleanroom QA — real-time verification that controlled conditions remain within specification.
Monitoring Requirements:
- Viable particles - Microbial contamination via air sampling, surface sampling, and personnel monitoring
- Non-viable particles - Continuous or periodic particle counting at sizes ≥0.5 μm and ≥5 μm
- Temperature and humidity - Continuous recording with acceptable ranges based on product/process requirements
- Differential pressure - FDA recommends continuous monitoring throughout each shift with frequent recording
Sampling locations must be selected based on risk assessment, targeting areas with highest contamination potential near sterile equipment, containers, and products. EU GMP Annex 1 requires justification of all monitoring locations within the Contamination Control Strategy.
Alert and Action Limits:
- Microbiological - FDA recommends action levels (e.g., 1 CFU/m³ for ISO 5) but permits alternative limits with scientific justification
- Particulate - ISO 14644-1 defines classification limits; Annex 1 specifies Grade A-D thresholds
- Limits should be established from baseline data collected during performance qualification
Facility Qualification and Validation
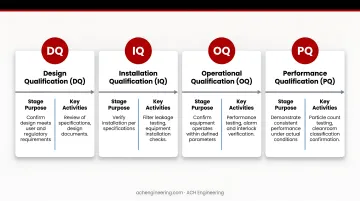
The DQ/IQ/OQ/PQ process demonstrates cleanroom compliance from design through operation:
| Stage | Purpose | Key Activities |
|---|---|---|
| Design Qualification (DQ) | Confirm design meets user and regulatory requirements | Review of specifications, design documents |
| Installation Qualification (IQ) | Verify installation per specifications | Filter leakage testing, equipment verification |
| Operational Qualification (OQ) | Confirm systems operate within defined parameters | Airflow measurement, pressure differentials, visualization studies |
| Performance Qualification (PQ) | Demonstrate consistent performance under real conditions | Microbial contamination testing, operational trials |
Requalification triggers include:
- Significant equipment changes or modifications
- Recurring environmental monitoring excursions
- Annual or biennial scheduled cycles
- Process changes affecting contamination risk
Standard Operating Procedures
Critical SOPs required for compliant cleanroom operations include:
- Gowning procedures - Step-by-step donning sequences with quality checkpoints
- Cleaning protocols - Frequencies, agents, methods, and acceptance criteria
- Material transfer - Procedures preventing contamination during material entry/exit
- Environmental monitoring - Sampling locations, frequencies, methods, and documentation
- Deviation management - Investigation requirements and CAPA initiation criteria
Personnel Training and Qualification
21 CFR 211.25 mandates that personnel have appropriate education, training, and experience for assigned functions. Training must cover:
- GMP principles and regulatory requirements
- Aseptic techniques and contamination sources
- Proper gowning and hygiene practices
- Environmental monitoring procedures
- Deviation reporting responsibilities
Qualification processes require documented competency assessments before personnel work independently, including written tests, observed performance evaluations, and media fill simulations for aseptic operations.
Corrective and Preventive Action (CAPA) System
Effective CAPA addresses deviations systematically — from root cause through verified resolution:
- Immediate containment actions to protect product quality
- Root cause analysis using structured methodologies (5 Whys, fishbone diagrams)
- Corrective actions addressing identified root causes
- Preventive actions eliminating potential future occurrences
- Effectiveness verification confirming the root cause is eliminated and excursions have not recurred
- Trend analysis identifying systemic weaknesses
Implementing Quality Assurance Protocols
Phased Implementation Approach
Successful QA program implementation follows a structured methodology:
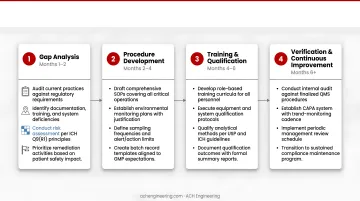
Phase 1: Gap Analysis (Months 1-2)
- Audit current practices against regulatory requirements
- Identify documentation, training, and system deficiencies
- Conduct risk assessment per ICH Q9(R1) principles
- Prioritize remediation activities based on patient safety impact
Phase 2: Procedure Development (Months 2-4)
- Draft comprehensive SOPs covering all critical operations
- Establish environmental monitoring plans with justified sampling locations
- Define alert and action limits based on cleanroom classification
- Create training curricula addressing identified gaps
Phase 3: Personnel Training (Months 3-5)
- Conduct classroom training on new procedures and GMP principles
- Perform hands-on qualification for critical operations
- Document training completion and competency assessments
- Establish ongoing training schedules
Phase 4: Pilot Testing (Months 4-5)
- Execute procedures on limited scale to identify practical issues
- Refine SOPs based on operator feedback
- Verify monitoring systems capture required data
- Test CAPA system with simulated deviations
Phase 5: Full Implementation (Month 6)
- Roll out complete QA program across all operations
- Initiate routine environmental monitoring
- Begin trend analysis and management review cycles
- Schedule first internal audit
Establishing Baseline Performance
Once full implementation is underway, initial qualification studies establish the baseline data used to set appropriate alert and action limits. Performance qualification typically runs 2-4 weeks — long enough to capture normal variability in environmental conditions and produce statistically valid limits.
Alert limits typically fall at 50-75% of action limits, providing early warning of adverse trends before specifications are exceeded.
Integration with Quality Management Systems
QA protocols must integrate directly with existing quality management systems, requiring cross-functional collaboration between operations, engineering, and quality teams. Regular management reviews should assess program effectiveness, review trending data, and allocate resources for continuous improvement.
Embedding QA requirements from the design phase — rather than retrofitting them — significantly reduces compliance risk. ACH Engineering's turnkey cleanroom solutions follow this approach: compliance requirements are built into facility infrastructure from the start, and modular environmental control systems are designed to adapt as regulatory standards evolve.
Monitoring, Testing, and Documentation Requirements
Routine Testing Requirements
Particle Counting:
- Continuous monitoring for Grade A zones during critical operations
- Periodic sampling (daily to weekly) for lower classifications
- Real-time trending to detect adverse patterns
- Documented investigation of excursions
Microbial Sampling:
- Air sampling - Active (volumetric) or passive (settle plates) methods
- Surface sampling - Contact plates or swabs for equipment and facility surfaces
- Personnel monitoring - Gown and glove sampling post-operation
HEPA Filter Integrity Testing:
- Performed at installation, after filter replacement, and at scheduled intervals (typically 6-12 months)
- Uses aerosol challenge to detect leaks in media, frames, and gaskets
- Follows IEST-RP-CC034.5 procedures
Airflow Visualization Studies:
- Smoke studies demonstrating unidirectional flow patterns
- Verification that airflow moves from clean to less clean areas
- Documentation of flow disruptions near critical operations
Documentation Requirements
Comprehensive documentation proves compliance during regulatory inspections:
- Master Batch Records — Full production documentation capturing environmental conditions at time of manufacturing
- Logbooks — Real-time entries for monitoring data, cleaning activities, and personnel access
- Deviation Reports — Root cause investigations of all excursions, with linked CAPA actions
- CAPA Records — Tracked from initiation through effectiveness verification
- Training Records — Individual histories with documented competency assessments
- Qualification/Validation Packages — Complete DQ/IQ/OQ/PQ protocols with executed test results

Documentation Retention Requirements
Pharmaceuticals (21 CFR 211.180):
- Minimum 1 year after batch expiration date
- For OTC drugs without expiration dates: 3 years after distribution
Medical Devices (21 CFR 820.180):
- Minimum 2 years from commercial release
- Or equivalent to device design and expected life
Electronic vs. Paper-Based Systems
Electronic systems offer several practical advantages:
- Real-time data logging with automated trending
- Searchable databases for rapid information retrieval
- Automated alerts when parameters exceed limits
- Audit trails documenting all data modifications
- Integration with quality management systems
Electronic records must comply with 21 CFR Part 11 (US) and Annex 11 (EU) regarding validation, security, and audit trail requirements. Canadian facilities should also reference Health Canada's guidance documents for equivalent data integrity expectations.
Paper systems remain acceptable but are prone to transcription errors, physical damage, and time-consuming trend analysis. Recent FDA warning letters have repeatedly cited data integrity failures tied to inadequate paper record controls — a risk that electronic systems largely eliminate.
Common Compliance Challenges and Solutions
Frequent Compliance Issues
Recent FDA enforcement data reveals consistent violation patterns:
Inadequate Environmental Monitoring Programs:
- Insufficient sampling locations failing to cover high-risk areas
- Monitoring frequencies not justified by risk assessment
- Failure to monitor adjacent non-classified rooms
- Solution: Conduct a comprehensive risk assessment identifying all potential contamination sources; justify monitoring plans in your Contamination Control Strategy; implement continuous monitoring for critical zones
Incomplete Documentation:
- Missing batch records or environmental monitoring data
- Inadequate deviation investigations without root cause analysis
- Training records lacking competency verification
- Solution: Implement electronic systems with automated data capture; establish document review procedures before batch release; conduct regular internal audits verifying documentation completeness
Insufficient Personnel Training:
- Personnel working without documented qualification
- Training programs not covering aseptic techniques adequately
- Lack of ongoing refresher training
- Solution: Develop comprehensive training curricula with hands-on qualification; require annual refresher training; document all training with competency assessments
Failure to Investigate Deviations:
- Excursions dismissed without thorough investigation
- Root causes not identified or addressed
- Preventive actions not implemented
- To address this: Establish clear investigation triggers; use structured root cause analysis methodologies; implement a CAPA system with effectiveness checks built in
Balancing Production Demands with Compliance
Equipment upgrades, process modifications, and facility expansions each introduce compliance risks — most of which are manageable with a structured change control process in place.
A robust change control procedure should:
- Assess the impact on existing validations and qualifications
- Determine requalification scope based on the magnitude of the change
- Update environmental monitoring plans for new equipment or layouts
- Revise SOPs to reflect modified procedures
- Retrain personnel on changes before implementation
- Document change rationale and approval in quality records

Facility design plays a role here too. Modular cleanroom designs offer a practical compliance advantage: prefabricated components can be reconfigured, expanded, or relocated without invalidating the broader validated environment — something traditional construction rarely accommodates as cleanly.
Frequently Asked Questions
What is the difference between ISO 14644 and FDA cleanroom standards?
ISO 14644 defines particle concentration limits and testing methodologies for air cleanliness classification. FDA regulations (21 CFR Parts 210, 211, and 820) cover broader GMP requirements — documentation, validation, quality systems, and sterility assurance. ISO sets the technical particle classification framework; FDA focuses on the quality systems that ensure product safety and efficacy.
How often should cleanroom quality assurance audits be conducted?
Most facilities conduct internal audits quarterly or semi-annually for critical systems, adjusting frequency based on risk assessment and past performance. 21 CFR 820.22 requires "periodic" audits without specifying exact intervals, while external regulatory audits typically occur annually or biennially. EU GMP Annex 1 requires environmental monitoring data review as part of the periodic Contamination Control Strategy review.
What documentation is required for cleanroom compliance?
Core documentation requirements include:
- SOPs covering all critical operations
- Environmental monitoring records (particle counts, microbial sampling, temperature/humidity/pressure logs)
- Equipment qualification and calibration records (DQ/IQ/OQ/PQ protocols)
- Personnel training records with competency assessments
- Deviation reports with root cause investigations and CAPA tracking
- Periodic audit reports documenting compliance verification
What are the most common cleanroom compliance violations?
FDA warning letters most frequently cite:
- Inadequate environmental monitoring (insufficient locations, frequencies, or failure to investigate excursions)
- Facility design failures (poor materials, rust, or particle board in controlled areas)
- Incomplete training documentation and improper gowning procedures
- Insufficient cleaning validation
- Failure to investigate discrepancies or batch failures per 21 CFR 211.192
Do I need a dedicated quality assurance team for my cleanroom?
The need depends on facility size, complexity, and regulatory requirements. Small operations with limited product lines may use part-time QA resources or consultants. Larger pharmaceutical or medical device facilities typically require dedicated QA personnel given the volume of documentation, monitoring data review, deviation investigations, and audit preparation. If your facility is subject to FDA inspection, a full-time QA staff member with regulatory expertise is rarely optional in practice.
How long does it take to implement a cleanroom QA program?
Most facilities complete a full QA program implementation in 3-6 months: gap analysis (1-2 months), procedure development (2-3 months), training (concurrent), system validation (1 month), and baseline monitoring (2-4 weeks). Facilities building from minimal QA infrastructure should plan for the longer end of that range.


