
Introduction
Pharmaceutical manufacturers face a critical challenge: contamination in production environments can compromise patient safety, trigger costly regulatory failures, and derail product launches. According to the FDA's inspection observations database, environmental control and documentation failures consistently rank among the top compliance citations, often resulting in warning letters and production halts.
The key to addressing these risks lies in proper cleanroom classification. Understanding the classification system—governed by ISO 14644-1 and EU GMP Annex 1—is essential for ensuring sterile production environments while optimizing facility costs.
These standards determine everything from HVAC design and filtration requirements to monitoring protocols and operational procedures. Yet many manufacturers over-specify cleanroom grades, driving construction costs from $150 per square foot to over $1,000 per square foot without meaningful safety improvements.
This article provides practical guidance for pharmaceutical manufacturers:
- Breakdown of ISO and EU GMP classification systems
- GMP compliance requirements for each cleanroom grade
- Critical design specifications and cost considerations
- How to select appropriate classifications without over-engineering
Key Takeaways
- Two classification systems govern pharmaceutical cleanrooms: ISO 14644-1 (Classes 1-9) and EU GMP (Grades A-D)
- Match your process to ISO Classes 5-8—sterile operations need tighter controls than non-sterile
- Meet GMP requirements through HEPA/ULPA filtration, environmental monitoring, and validated procedures
- Risk-based classification selection prevents costly over-specification while ensuring patient safety and regulatory approval
Understanding Pharmaceutical Cleanroom Classification Systems
Cleanroom classification measures air cleanliness based on particle count per cubic meter at specified sizes (primarily 0.5 µm and 5.0 µm).
Classification is not a design specification but an operational requirement that drives HVAC design, filtration needs, and monitoring protocols.
The classification determines maximum allowable particle concentration. Lower ISO class numbers indicate cleaner environments—ISO Class 5 is significantly cleaner than ISO Class 7, requiring exponentially more air changes and filtration coverage.
ISO 14644-1 Classification Framework
ISO 14644-1:2015 establishes the global standard for cleanroom classification using a logarithmic scale. Each step in class number represents a tenfold change in allowable particle concentration.
ISO Cleanroom Classification Limits
| ISO Class | Max Particles/m³ (≥0.5 µm) | Max Particles/m³ (≥5.0 µm) | Typical Pharmaceutical Application |
|---|---|---|---|
| ISO 5 | 3,520 | 29 | Aseptic processing (filling zones) |
| ISO 6 | 35,200 | 293 | Support zones for ISO 5 areas |
| ISO 7 | 352,000 | 2,930 | Background areas, preparation rooms |
| ISO 8 | 3,520,000 | 29,300 | Component preparation, packaging |
| ISO 9 | 35,200,000 | 293,000 | Controlled non-classified areas |
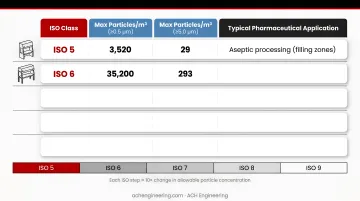
Pharmaceutical manufacturers most commonly use ISO Classes 5-8:
- ISO 5: Critical aseptic processing zones (filling, vial handling)
- ISO 6: Direct support areas surrounding ISO 5 environments
- ISO 7-8: Preparation rooms and less critical operations
EU GMP Grade System
The EU GMP Annex 1 (2022 revision) defines four grades (A, B, C, D) specifically for sterile medicinal product manufacture. Unlike ISO 14644-1, EU GMP distinguishes between "at rest" (no personnel present) and "in operation" (normal production activity) states.
EU GMP Grades and ISO Equivalents
| Grade | At Rest (≥0.5 µm) | In Operation (≥0.5 µm) | ISO Equivalent (At Rest) | Application |
|---|---|---|---|---|
| Grade A | 3,520 | 3,520 | ISO 5 | Critical zone (filling, aseptic connections) |
| Grade B | 3,520 | 352,000 | ISO 5 | Background for Grade A in aseptic preparation |
| Grade C | 352,000 | 3,520,000 | ISO 7 | Less critical stages, terminal sterilization prep |
| Grade D | 3,520,000 | Not predetermined | ISO 8 | Component handling, initial preparation |
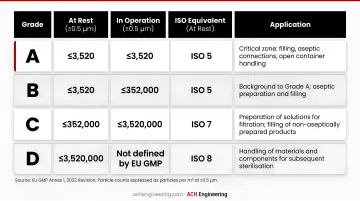
Key distinction: Grade A requires unidirectional (laminar) airflow to continuously sweep particles away from critical processing points. Grades B-D use non-unidirectional (turbulent) airflow with sufficient air changes to maintain particle limits.
Legacy Standards and Transition Considerations
While current regulations reference ISO and EU GMP standards, many existing facilities were built under older classification systems.
FED-STD-209E was the dominant US cleanroom standard until its official cancellation in November 2001. Despite being obsolete for over two decades, legacy terminology like "Class 100" or "Class 10,000" still appears in older facilities and documentation.
Legacy to ISO Conversion
| FED-STD-209E Class | ISO 14644-1 Equivalent | Particles/ft³ (≥0.5 µm) |
|---|---|---|
| Class 100 | ISO 5 | 100 |
| Class 1,000 | ISO 6 | 1,000 |
| Class 10,000 | ISO 7 | 10,000 |
| Class 100,000 | ISO 8 | 100,000 |
Transition guidance for facilities using legacy classifications:
- Update all documentation, SOPs, and specifications to ISO 14644-1 classifications
- Revise master plans and validation protocols to reflect current standards
- Retrain personnel on ISO terminology and requirements
- Note: Regulatory submissions using legacy terminology may face scrutiny or rejection
GMP Compliance Requirements for Pharmaceutical Cleanrooms
Regulatory Framework Overview
The FDA (United States), EMA (European Union), and other regional authorities enforce Good Manufacturing Practice (GMP) to ensure pharmaceutical products are consistently produced and controlled according to quality standards.
GMP guidelines prescribe how cleanrooms must be designed, operated, validated, and monitored—extending far beyond particle counts. Pharmaceutical manufacturers must comply with both ISO cleanroom standards (which define air cleanliness) and GMP operational requirements (which govern processes, documentation, and quality systems).
The FDA's 21 CFR Part 211 establishes US requirements for finished pharmaceuticals, while EU GMP Annex 1 provides European sterile manufacturing standards. Both frameworks require environmental control systems, validated processes, and comprehensive documentation.
Critical GMP Elements for Cleanrooms
Pharmaceutical cleanrooms must meet specific GMP requirements:
- Qualification (IQ/OQ/PQ): Installation Qualification verifies equipment is installed correctly, Operational Qualification confirms it operates within parameters, and Performance Qualification proves consistent performance under actual conditions
- Environmental monitoring: Continuous or periodic sampling of viable and non-viable particles, temperature, humidity, and pressure differentials
- Personnel training: Documented training programs covering gowning procedures, aseptic techniques, and contamination control behaviors
- Cleaning validation: Protocols proving cleaning procedures effectively remove residues and reduce microbial contamination to acceptable levels
- Change control: Formal systems for evaluating, approving, and documenting modifications to facilities, equipment, or processes

Qualification proves the facility and equipment are designed and installed correctly, while validation proves the manufacturing process consistently produces products meeting quality standards.
Full GMP qualification of a new pharmaceutical cleanroom typically requires 3-6 months and represents 15-25% of total project costs, depending on complexity and classification.
Documentation and Traceability
Comprehensive documentation forms the foundation of GMP compliance. Critical documentation elements include:
- Standard Operating Procedures (SOPs): Written instructions for all critical operations, including cleaning, gowning, environmental monitoring, and equipment operation
- Batch records: Complete documentation of each production batch, including environmental conditions during manufacturing
- Environmental monitoring logs: Continuous or periodic records of particle counts, microbial levels, temperature, humidity, and pressure
- Deviation reports: Documentation of any excursions from established parameters, including investigation and corrective actions
Electronic monitoring systems (Building Management Systems and Environmental Monitoring Systems) maintain continuous compliance records, generate real-time alerts for excursions, and provide audit trails required during regulatory inspections.
Documentation failures consistently appear among top FDA inspection findings. Missing records, inadequate investigations of environmental excursions, and poorly maintained SOPs trigger warning letters and regulatory actions.
Critical Design Features for GMP-Compliant Cleanrooms
HVAC and Air Filtration Systems
Filter selection depends on required classification:
- HEPA (High-Efficiency Particulate Air): H14 filters achieve ≥99.995% efficiency at 0.1-0.2 µm (Most Penetrating Particle Size). Standard for pharmaceutical cleanrooms ISO 5-8
- ULPA (Ultra-Low Penetration Air): U15 filters achieve ≥99.9995% efficiency at 0.12 µm. Required for specialized applications demanding extreme cleanliness
Airflow patterns differ by classification:
- Unidirectional (laminar) flow: Required for Grade A (ISO 5) critical zones. Air flows in parallel streams at 0.36-0.54 m/s (70-105 fpm), continuously sweeping particles away from product contact surfaces
- Non-unidirectional (turbulent) flow: Used for Grades B-D (ISO 5-8 at rest). Air enters through HEPA filters in ceiling, mixes throughout room, and exits through low-level returns
Air change rates (ACH) determine how quickly air is replaced:
| ISO Class | Typical ACH | Recovery Time |
|---|---|---|
| ISO 5 (Grade A) | 300-480 | <5 minutes |
| ISO 7 (Grade B/C) | 40-60 | 15-20 minutes |
| ISO 8 (Grade D) | 20-40 | 15-20 minutes |

Higher air change rates mean faster particle removal but exponentially higher energy costs and HVAC system complexity.
Pressure Differentials and Cascade Control
Positive pressure prevents contamination entry from adjacent areas. Cleaner areas are maintained at higher pressure than less-clean areas, creating outward airflow through any gaps or openings.
Typical pressure differential specifications:
- Minimum 10-15 Pascals (Pa) between adjacent rooms of different grades
- Minimum 5 Pa between rooms of the same classification when separation is needed
Contamination barriers maintain pressure cascades:
- Airlocks: Transition zones between areas of different classifications, with separate doors that prevent simultaneous opening
- Pass-throughs: Sealed chambers for material transfer between classified areas without personnel movement
- Gowning rooms: Controlled spaces where personnel don appropriate cleanroom garments before entering higher-grade areas
Automated Building Management Systems continuously monitor pressure differentials and trigger alarms when values fall below acceptable limits.
Material Selection and Surface Requirements
GMP-compliant materials must be:
- Non-shedding: Materials that don't release particles or fibers into the environment
- Impervious: Smooth, sealed surfaces that don't absorb moisture or harbor microorganisms
- Chemical-resistant: Able to withstand repeated cleaning with disinfectants and sanitizers
Common GMP-compliant materials include:
- Wall panels: Non-porous polymer-coated panels with sealed joints
- Flooring: Seamless epoxy or polyurethane with coved edges
- Fixtures: Stainless steel (typically 316L grade) for equipment, furniture, and hardware
**Coved corners and sealed joints** eliminate 90-degree angles where particles accumulate. Seamless wall-floor and wall-ceiling transitions facilitate cleaning validation and prevent contamination harborage.
Modular vs. Traditional Cleanroom Construction
These material requirements directly influence construction approach. Two primary methods exist for pharmaceutical cleanroom projects.
Traditional stick-built construction involves on-site fabrication of walls, ceilings, and systems. Timeline: 15-30 months from design to commissioning.
This approach offers unlimited customization. However, it introduces weather delays, quality variability, and extended downtime.
Modular prefabricated construction uses factory-manufactured components assembled on-site. Timeline: Design and fabrication in weeks, on-site installation in days to weeks.
ACH Engineering's modular cleanroom systems offer several advantages for pharmaceutical applications:
- Faster deployment: Factory testing and quality control reduce on-site installation time and accelerate commissioning
- Quality consistency: Controlled manufacturing environment ensures components meet specifications
- Future flexibility: Modular systems can be expanded, reconfigured, or relocated as production needs change
- Full GMP compliance: Prefabricated components include cGMP-compliant coving, HEPA filtration integration, and validated materials
These advantages translate directly to project success. Modular construction typically reduces project timelines by 40-60% compared to traditional methods, allowing pharmaceutical manufacturers to begin production sooner while maintaining full regulatory compliance.
Environmental Monitoring and Control Systems
Continuous environmental monitoring is a GMP requirement, not optional. These programs verify cleanrooms remain in control during operations and detect contamination events before product quality is affected.
Particle Counting and Viable Monitoring
Non-viable (inert) particle monitoring uses automated particle counters at critical locations:
- Sample air continuously or periodically
- Count particles at 0.5 µm and 5.0 µm size ranges
- Generate real-time alerts when counts exceed action levels
- Grade A zones require continuous monitoring during operations
Viable (microbial) monitoring detects living microorganisms through:
- Active air sampling: Volumetric samplers draw air across agar plates to capture microorganisms
- Settle plates: Open agar plates placed in the environment to capture settling microorganisms
- Surface swabs: Direct sampling of equipment and surfaces to detect contamination
Typical monitoring frequencies (EU GMP):
| Grade | Particle Monitoring | Viable Monitoring |
|---|---|---|
| Grade A | Continuous during operations | Frequent (every fill/batch) |
| Grade B | Frequent during operations | Frequent (daily-weekly) |
| Grade C | Periodic (daily-weekly) | Periodic (weekly-monthly) |
| Grade D | Periodic (weekly-monthly) | Periodic (monthly) |

Temperature, Humidity, and Pressure Monitoring
Beyond particle and microbial monitoring, environmental parameters directly impact both product quality and contamination control.
Temperature control maintains conditions typically between 18-22°C or 20-24°C (pharmaceutical standards use Celsius globally):
- Affects personnel comfort and reduces particle generation from excessive movement
- Influences product stability during processing
- Prevents condensation that can harbor microbial growth
Humidity control (typically 35-55% RH):
- Low humidity (<30%) increases static electricity and particle generation
- High humidity (>60%) promotes microbial growth and affects hygroscopic products
- Must be validated for specific product requirements
Pressure monitoring:
- Continuous measurement of pressure differentials between adjacent rooms
- Automated alarms when differentials fall below minimum thresholds
- Critical for maintaining contamination control strategy
Building Management Systems (BMS) integrate all environmental parameters to provide:
- Real-time monitoring dashboards
- Automated alarming for excursions
- Historical trending for investigation and validation
- Electronic records for regulatory compliance
Common Compliance Challenges and How to Avoid Them
Even well-designed cleanrooms face compliance issues. Most stem from operations rather than design flaws.
Personnel as Primary Contamination Source
Humans are the largest contamination source in cleanrooms. Research shows that personnel generate approximately 70% of airborne particles in occupied cleanrooms.
A single person emits 100,000-1,000,000 particles per minute depending on activity level.
Contamination control strategies:
- Proper gowning procedures: Multi-stage gowning in dedicated rooms with documented procedures and regular competency assessments
- Behavior protocols: Minimize unnecessary movement, avoid touching face or hair, maintain proper gown integrity
- Personnel flow patterns: Unidirectional flow from clean to dirty areas, separate entry and exit paths
- Training and qualification: Initial and ongoing training with documented competency verification
Poor gowning technique or inappropriate behavior can compromise even the most sophisticated cleanroom design.
Beyond personnel management, design decisions themselves create significant compliance risks—particularly when cost considerations aren't balanced with actual regulatory requirements.
Over-Classification and Cost Implications
Specifying ISO Class 5 when ISO Class 7 meets regulatory requirements drives unnecessary costs. Construction costs escalate dramatically with each classification step:
- ISO 8: $75-150 per square foot (20-40 ACH, 4-5% HEPA coverage)
- ISO 7: $200-400 per square foot (40-60 ACH, 15-20% HEPA coverage)
- ISO 5: $1,000+ per square foot (300-480 ACH, 60-70% HEPA coverage)
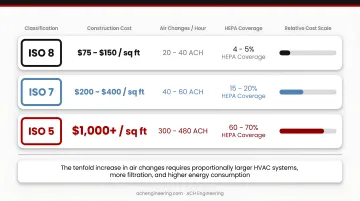
The tenfold increase in air changes requires proportionally larger HVAC systems, more filtration, and higher energy consumption. Yet it provides no additional safety if the lower classification already meets regulatory requirements.
Risk-based classification selection guidance:
- Match classification to product sterility requirements and processing stage
- Consult FDA guidance documents and EU GMP Annex 1 for minimum requirements
- Consider terminal sterilization vs. aseptic processing pathways
- Design for appropriate classification, not ideal "best possible" cleanliness
Inadequate Cleaning Validation
Effective cleaning validation prevents contamination events and audit findings.
Common failures include:
- Using non-validated disinfectants that aren't effective against target organisms
- Inconsistent cleaning procedures without documented frequencies or methods
- Lack of surface monitoring to verify cleaning effectiveness
- Failure to establish acceptance criteria for residues and microbial levels
Recommended approach:
- Establish cleaning validation protocols proving effectiveness against relevant microorganisms
- Use multiple disinfectants with different mechanisms of action
- Implement routine surface monitoring (swabs, contact plates)
- Document all cleaning activities with batch-specific records
- Investigate and address any environmental excursions promptly
Selecting the Right Classification for Your Pharmaceutical Process
Classification selection is a risk-based decision balancing patient safety, regulatory requirements, and operational costs.
Classification by Product Type and Process Stage
| Product Type | Process Stage | ISO Classification | EU GMP Grade |
|---|---|---|---|
| Sterile injectables (aseptic) | Critical zone (filling, stopper placement) | ISO Class 5 | Grade A with unidirectional airflow |
| Background environment | ISO Class 7 | Grade B at rest, Grade C in operation | |
| Preparation areas | ISO Class 7-8 | Grade C-D | |
| Sterile products (terminal sterilization) | Filling operations | ISO Class 7 | Grade C in operation |
| Preparation and component handling | ISO Class 8 | Grade D | |
| Non-sterile solid dosage | Manufacturing areas | ISO Class 8 or CNC | Focus on cross-contamination prevention |
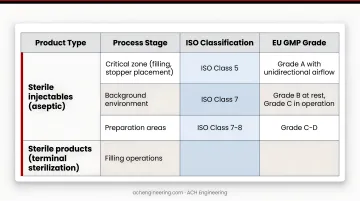
FDA guidance on microbiological quality for non-sterile products and EU GMP Annex 1 specify classification requirements for different product types and processing stages.
Regulatory Expectations and Inspection Readiness
Early consultation with regulatory guidelines prevents costly redesigns. Key resources to review include:
- FDA 21 CFR Part 211 and relevant guidance documents for US markets
- EU GMP Annex 1 when targeting European distribution
- PIC/S guidelines for international harmonization across multiple regions
Translating these requirements into compliant facility designs requires specialized expertise. Design-build partners with pharmaceutical project experience—such as ACH Engineering, an ISPE member who provides FDA cleanroom design services—bring familiarity with both technical standards and regulatory expectations that help avoid compliance gaps during design.
Pre-approval strategies:
- Mock audits conducted before regulatory submission identify gaps early
- Documentation and procedure reviews address deficiencies proactively
- Thorough validation of environmental monitoring and control systems
- Comprehensive qualification documentation (IQ/OQ/PQ) prepared for inspection
Future Flexibility and Scalability Considerations
Design for potential classification upgrades without major reconstruction. Building in flexibility now prevents costly retrofits later.
Key design strategies include:
- Install HVAC capacity for future higher air change rates
- Provide infrastructure for additional HEPA filters
- Use modular construction that allows reconfiguration
- Plan for additional monitoring points and equipment
Modular cleanroom systems allow easier modification as product lines or regulations change. Components can be relocated, expanded, or upgraded without complete facility reconstruction. This approach protects long-term capital investment while maintaining compliance flexibility.
Conclusion
Pharmaceutical cleanroom classification integrates particle count standards with comprehensive systems covering design, operation, monitoring, and documentation.
Proper classification selection and GMP compliance are non-negotiable for patient safety and regulatory approval, yet over-specification drives unnecessary costs without meaningful benefit.
Success requires understanding the relationship between ISO 14644-1 classifications and EU GMP grades, selecting appropriate classifications based on product type and processing stage, and implementing comprehensive environmental control and monitoring systems.
Achieving regulatory readiness demands experienced partners who can navigate these requirements. ACH Engineering provides modular, GMP-compliant cleanroom solutions for pharmaceutical manufacturers across North America, with turnkey delivery from design through qualification, factory-tested components, and flexibility for future expansion.
Frequently Asked Questions
What are the requirements for a pharmacy cleanroom?
Pharmacy cleanrooms require appropriate ISO classification (typically ISO 7-8 for compounding), HEPA filtration with positive pressure, continuous environmental monitoring, documented gowning procedures, and validated cleaning protocols. USP 797 and 800 provide specific compounding pharmacy guidance per GMP requirements.
What is a clean area in the pharmaceutical industry?
A clean area is a defined space with controlled airborne particulate and microbial contamination levels, classified by ISO 14644-1 (Classes 1-9) or EU GMP grades (A-D). These environments protect pharmaceutical manufacturing where contamination could compromise product safety, efficacy, or stability.
What is the ISO standard for the pharmaceutical industry?
ISO 14644-1 defines cleanroom classifications (Classes 1-9) based on particle concentrations. Pharmaceutical manufacturers must also comply with GMP regulations like FDA 21 CFR Part 211 (US) and EU GMP Annex 1 (Europe), which specify operational, validation, and monitoring requirements beyond particle counts.


