
Introduction
Pharmaceutical elastomeric closures—rubber stoppers for vials, syringes, and cartridges—directly contact sterile drug products throughout their shelf life. A single contamination event during stopper washing, inspection, or packaging can compromise an entire batch, trigger recalls, and jeopardize patient safety.
ISO cleanroom standards provide the critical framework for contamination control in stopper manufacturing, defining allowable particle concentrations and environmental conditions that protect product sterility.
Regulatory agencies including the FDA, EMA, and PMDA require demonstrated contamination control through validated cleanroom environments for components contacting sterile drugs. The consequences of non-compliance are well-documented: 0.45% of pharmaceutical recalls between 2012–2021 were directly linked to endotoxin contamination, often traceable to component preparation failures, and 686 microbial contamination recalls were recorded in the US from 2012–2023. Getting cleanroom classification right from the start is what separates compliant operations from costly ones.
This guide covers ISO 14644 classifications, GMP requirements, cleanroom design considerations, and compliance strategies specifically for pharmaceutical stopper manufacturing.
Key Takeaways
- ISO 7 or ISO 8 cleanrooms are typically required depending on sterilization method and drug application
- EU GMP Grade C (ISO 7 at rest) is mandatory for stopper preparation before terminal sterilization
- Particle limits differ by class: ISO 7 caps at 352,000 particles ≥0.5 µm/m³; ISO 8 allows up to 3,520,000 particles ≥0.5 µm/m³
- HEPA filtration, pressure cascades, and environmental monitoring together prevent particulate, microbial, and endotoxin contamination
- ISO 14644-2 validation requires testing every 6–12 months and after any major HVAC modifications
Why Cleanroom Standards Matter for Stopper Manufacturing
Direct Contact with Sterile Products
Pharmaceutical stoppers serve as primary packaging components that seal vials, syringes, and cartridges containing injectable drugs. Unlike secondary packaging, these elastomeric closures remain in direct contact with the drug product from filling through administration, making them critical to sterility assurance.
Contamination Risks Specific to Stoppers
Stopper manufacturing presents unique contamination challenges:
- Elastomeric materials shed particles through friction during handling, washing, and inspection — a risk unique to rubber-based closures
- Uncontrolled processing environments introduce microbial contamination that terminal sterilization may not fully eliminate
- Bacterial endotoxins survive sterilization cycles and can trigger severe pyrogenic reactions in patients
- Chemical contaminants from poorly controlled environments migrate into drug products as extractables and leachables
FDA guidance on container closure systems requires that elastomeric components be processed to remove pyrogenic properties and protect products from contamination.
Regulatory Requirements
Global regulatory agencies mandate contamination control through validated cleanroom environments:
- FDA — Requires packaging systems to be "suitable for intended use" with validated washing and depyrogenation processes achieving >3 log reduction in endotoxins
- EMA/EU GMP Annex 1 — Explicitly mandates Grade C (ISO 7 at rest) for component preparation for terminally sterilized products
- PMDA (Japan) — Requires ISO Class 5 in filling and sealing areas where stoppers contact sterile products
- ISO 13485 / cGMP — Cleanroom classification requirements integrate directly with medical device and pharmaceutical quality management systems, forming the backbone of a site's contamination control program

Consequences of Inadequate Standards
Failures in cleanroom compliance result in:
- Product recalls affecting thousands of units
- FDA warning letters and consent decrees
- Batch rejections costing millions in lost revenue
- Patient safety incidents including pyrogenic reactions
- Regulatory approval delays for new drug applications
ISO 14644 Cleanroom Classifications Explained
The International Standard
ISO 14644-1:2015 defines cleanroom air cleanliness based on airborne particle concentration per cubic meter. It serves as the global foundation for pharmaceutical cleanroom design and is the standard all current regulatory filings must reference.
ISO Classification Scale
The standard defines nine classes from ISO 1 (cleanest) to ISO 9. For stopper manufacturing, ISO 5 through ISO 8 are most relevant:
Particle Concentration Limits
| ISO Class | Max Particles/m³ (≥0.5 µm) | Max Particles/m³ (≥5.0 µm) | Typical Stopper Application |
|---|---|---|---|
| ISO 5 | 3,520 | 29* | Aseptic filling, RTU stopper handling |
| ISO 6 | 35,200 | 293 | Support zones (less common) |
| ISO 7 | 352,000 | 2,930 | Siliconization, component preparation |
| ISO 8 | 3,520,000 | 29,300 | Washing, initial handling |
Note: ISO 14644-1:2015 does not specify limits for ≥5.0 µm particles in ISO 5, though pharmaceutical applications often reference 29 particles/m³ to align with EU GMP Annex 1 Grade B requirements.
At Rest vs. In Operation States
ISO 14644-1 distinguishes between two critical operating conditions:
- At rest - Installation complete with equipment operating but no personnel present
- In operation - Normal functioning with specified personnel working in the space
EU GMP Annex 1 requires Grade B zones to meet ISO 5 at rest but permits ISO 7 in operation — a two-class gap that directly reflects how personnel activity drives particle counts. Both conditions must be tested and qualified during cleanroom validation.
Air Change Rates and HEPA Filtration
While ISO 14644-1 defines particle count results, industry baselines provide necessary air change rates (ACH):
- ISO 5 - 240-360 ACH (unidirectional flow)
- ISO 6 - 90-180 ACH
- ISO 7 - 30-60 ACH
- ISO 8 - 10-25 ACH
Achieving these air change rates requires HEPA filtration sized to the classification:
- H14 filters (99.995% efficiency) for ISO 5 zones
- H13 filters (99.95% efficiency) for ISO 7-8 supply air
Legacy Federal Standard 209E Equivalents
For reference to older documentation:
- Class 100 = ISO 5
- Class 10,000 = ISO 7
- Class 100,000 = ISO 8
All current regulatory filings should reference ISO classifications exclusively, as FS 209E was withdrawn in 2001.
Selecting the Right ISO Class for Stopper Production
Three Key Decision Factors
ISO class selection depends on:
- Stopper application - Injectable drugs vs. diagnostic devices
- Sterilization method - Terminal sterilization vs. aseptic processing
- Regulatory market - FDA, EMA, or PMDA requirements
ISO 8 for Terminally Sterilized Products
Use ISO 8 for stoppers destined for products undergoing terminal sterilization — steam, radiation, or ethylene oxide — where the sterilization cycle itself controls final bioburden.
When to use: Products with validated terminal sterilization cycles
Rationale: EU GMP Annex 1 permits Grade D (ISO 8) for initial washing and handling of components that will undergo validated terminal sterilization cycles. The final sterilization cycle achieves a Sterility Assurance Level (SAL) of ≤10⁻⁶, compensating for higher initial bioburden.
Operations: Initial washing, drying, basic handling
These requirements escalate significantly for products that cannot rely on terminal sterilization.
ISO 7 for Aseptically Filled Products
Use ISO 7 when stoppers are intended for aseptically filled products or when lower initial bioburden is required before sterilization.
When to use: Aseptically filled products or high-risk microbial contamination scenarios
Rationale: Annex 1 requires Grade C (ISO 7 at rest, ISO 8 in operation) for component preparation areas at high risk of microbial contamination. Unlike terminal sterilization, aseptic processing offers no downstream kill step to correct upstream contamination.
Operations: Siliconization, depyrogenation, sterilization preparation, final inspection
The strictest requirements apply where stoppers enter pre-sterilized, ready-to-use formats.
ISO 5 or ISO 6 for Critical Operations
Ready-to-use (RTU) stoppers packaged in nest-and-tub systems for aseptic filling lines represent the highest-risk operation. Pre-sterilized stoppers transferred into Grade A filling zones require Grade A/B environments to maintain sterile barrier integrity — Japanese PMDA guidance mandates ISO Class 5 specifically for filling and sealing areas. Applicable operations include RTU packaging, aseptic stopper bowl loading, and all direct product contact zones.
Risk-Based Decision Criteria
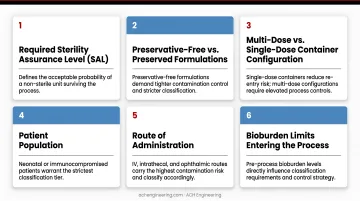
Consider these factors when selecting your classification:
- Required Sterility Assurance Level (SAL) for the final product
- Preservative-free vs. preserved formulations (preservative-free demands tighter control)
- Multi-dose vs. single-dose container configuration
- Patient population — neonatal or immunocompromised patients warrant stricter classification
- Route of administration — IV, intrathecal, and ophthalmic routes carry the highest contamination risk
- Bioburden limits entering the sterilization cycle, including D-value, Z-value, and whether an overkill or bioburden-based approach applies
GMP Requirements for Pharmaceutical Stoppers
EU GMP Annex 1 Classifications
The revised Annex 1 (2022) provides specific grade assignments for stopper operations:
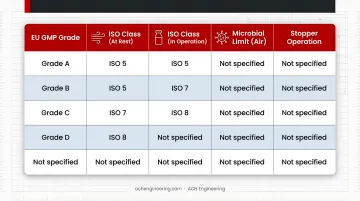
The table below maps each EU GMP grade to its corresponding ISO class, microbial limits, and typical stopper operation:
| EU GMP Grade | ISO Class (At Rest) | ISO Class (In Operation) | Microbial Limit (Air) | Stopper Operation |
|---|---|---|---|---|
| Grade A | ISO 5 | ISO 5 | <1 CFU/m³ | Aseptic stoppering, RTU transfer |
| Grade B | ISO 5 | ISO 7 | 10 CFU/m³ | Background for Grade A |
| Grade C | ISO 7 | ISO 8 | 100 CFU/m³ | Siliconization, depyrogenation |
| Grade D | ISO 8 | Not defined | 200 CFU/m³ | Washing, initial handling |
Grade C Requirements for Component Preparation
Annex 1 Section 4.14 states: "Preparation of components (stoppers) should be performed in at least a Grade D environment. If there is a high risk of microbial contamination, operations must occur in Grade C."
Grade C applications:
- Final washing before sterilization
- Siliconization operations
- Depyrogenation processes
- Final inspection and sorting
Grade D Acceptability
Where contamination risk is lower or validated terminal sterilization follows, Grade D (ISO 8) is sufficient. Acceptable operations include:
- Initial washing steps
- Component receiving and unpacking
- Handling after cleaning but before final preparation
- Operations where validated terminal sterilization follows
FDA Approach: Suitability Over Rigid Classes
While EU GMP Annex 1 assigns specific grade requirements by operation, FDA's 2015 guidance takes a different position — emphasizing "suitability" and validation over fixed classifications. The agency requires:
- Demonstration that packaging systems are "suitable for intended use"
- Validation of washing and depyrogenation processes (>3 log endotoxin reduction)
- Environmental monitoring data supporting contamination control
- Risk assessment justifying chosen controls
In practice, this means a facility can justify a higher or lower classification than Annex 1 prescribes — provided the validation data and risk assessment clearly support that decision.
Cleanroom Design Considerations for Stopper Manufacturing
Critical Design Elements
Airflow Patterns:
- Unidirectional flow (laminar) for Grade A zones at 0.36-0.54 m/s velocity
- Turbulent mixing ventilation acceptable for Grades C and D
- Air change rates matched to particle generation from equipment
Pressure Cascades:
- Maintain 10-15 Pascal (Pa) differential between adjacent grades
- Direction: Grade A → Grade B → Grade C → Grade D → unclassified areas
- Continuous monitoring with alarms for deviations
Airlocks and Material Transfer:
- Personnel airlocks with interlocked doors
- Material pass-throughs with HEPA filtration
- Rapid transfer ports for RTU stopper introduction
Material Flow Requirements
Design stopper processing workflows to prevent cross-contamination:
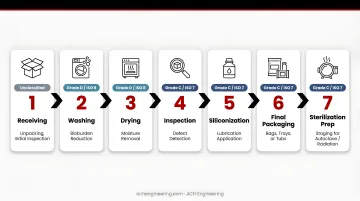
Sequential Flow:
- Receiving (unclassified) → unpacking, initial inspection
- Washing (Grade D/ISO 8) → bioburden reduction
- Drying (Grade D/ISO 8) → moisture removal
- Inspection (Grade C/ISO 7) → defect detection
- Siliconization (Grade C/ISO 7) → lubrication application
- Final Packaging (Grade C/ISO 7) → bags, trays, or tubs
- Sterilization Prep (Grade C/ISO 7) → staging for autoclave/radiation
Contamination Prevention:
- One-way material flow without backtracking
- Separate clean and dirty corridors
- Dedicated equipment for each zone
- No direct connection between Grade C and unclassified areas
Turnkey Solutions from ACH Engineering
Getting the zoning, airflow, and material flow right from the start requires coordinating a lot of interdependent systems. ACH Engineering offers turnkey cleanroom solutions that handle this coordination end-to-end — from initial classification zoning through validation — so pharmaceutical manufacturers don't have to manage multiple vendors across critical design decisions.
Their cleanroom design and build scope covers:
- Design and zoning: ISO classification mapping, pressure cascade calculations, HVAC system sizing, and material flow layout
- Modular construction: GMP-compliant wall panels (HPL, powder-coated galvanized iron, u-PVC) with seamless coving, flush-design doors, and walkable ceilings
- HVAC and environmental control: H13/H14 HEPA filtration, controlled pressurization, temperature (18–24°C), and humidity (30–60% RH)
- Qualification and documentation: ISO 14644-2 testing at rest and in operation, environmental monitoring integration, and regulatory submission packages
ACH serves pharmaceutical, biotech, and medical device cleanroom clients across Canada and North America, with projects built to meet FDA, EMA, and cGMP requirements.
Monitoring and Maintaining Compliance
Environmental Monitoring Requirements
Effective cleanroom compliance starts with a structured monitoring program covering particles, viable organisms, pressure, and environmental conditions.
Non-Viable Particle Counting:
- Grade A: Continuous monitoring during operations
- Grade C/D: Risk-based frequency (weekly to monthly)
- Sampling locations at critical process points and return air grilles
- Alert and action limits based on classification
Viable Monitoring:
- Air sampling: active (volumetric) and passive (settle plates)
- Surface sampling: contact plates or swabs on equipment and work surfaces
- Personnel monitoring: gown and glove sampling after operations
- Microbial limits: Grade A <1 CFU/m³, Grade C <100 CFU/m³, Grade D <200 CFU/m³
Pressure Differential Monitoring:
- Continuous monitoring with data logging
- Alarm systems for deviations below 10 Pa
- Verification during shift changes and after door openings
Temperature and Humidity Control:
- Continuous monitoring in all classified areas
- Typical ranges: 18–24°C, 30–60% RH
- Tighter controls required for specific stopper materials or processes
Validation Protocols
Beyond routine monitoring, formal validation testing confirms that your cleanroom consistently meets its classified performance levels — both at startup and throughout its operational life.
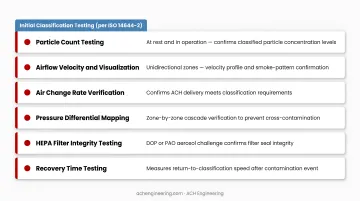
Initial Classification Testing (per ISO 14644-2):
- Particle count testing at rest and in operation
- Airflow velocity and visualization (for unidirectional zones)
- Air change rate verification
- Pressure differential mapping
- HEPA filter integrity testing (DOP or PAO challenge)
- Recovery time testing (cleanup period <20 minutes recommended)
Ongoing Monitoring Frequencies:
- Grade A/B: Every 6 months per Annex 1
- Grade C/D: Every 12 months per Annex 1
- After significant HVAC modifications
- Following contamination events or excursions
Requalification Intervals:
- EU GMP Annex 1 mandates more frequent testing than ISO 14644-2's risk-based approach
- Additional testing after filter changes, facility modifications, or process changes
- Trending of monitoring data to identify degradation before failure
Personnel Qualification Requirements
Equipment and environmental controls only go so far. Personnel behavior and hygiene are equally critical — and among the most common sources of contamination in stopper manufacturing environments.
Gowning Protocols:
- Grade D: Lab coats, hair covers, shoe covers
- Grade C: Full cleanroom suits, gloves, face masks
- Grade A/B: Sterile gowns, sterile gloves, goggles, breathing masks
Hygiene Controls:
- Hand washing and sanitization before gowning
- No cosmetics, jewelry, or personal items in classified areas
- Health monitoring programs (no personnel with active infections)
Behavioral Training:
- Minimize movements and talking in critical zones
- Proper material handling to reduce particle generation
- Understanding of contamination risks and control measures
- Annual retraining and competency assessment
Common Compliance Challenges and Solutions
Particle Count Excursions During Production
Washing, tumbling, and inspection equipment often generate particles that exceed classification limits — a persistent headache in stopper manufacturing lines.
- Enclose high-particle-generating equipment in mini-environments with dedicated HEPA filtration
- Increase local air change rates around problem equipment
- Modify processes to reduce mechanical stress on stoppers
- Schedule particle-intensive operations during non-critical periods with enhanced monitoring
Inadequate Pressure Differentials
Door openings, HVAC imbalances, and filter loading are the most common culprits when pressure drops fall below the 10 Pa minimum.
- Install automated pressure monitoring with real-time alarms
- Use interlocked airlocks to prevent simultaneous door openings
- Schedule HEPA filter changes based on pressure drop trends rather than fixed intervals
- Add makeup air systems to maintain positive pressure during high-traffic periods
- Commission a dedicated differential pressure map to identify weak points early
Improper Gowning Compliance
Personnel gowning errors account for roughly 80% of viable contamination in cleanrooms — making this the single highest-impact compliance area to get right.

- Set up video-monitored gowning areas with supervisor verification
- Run gowning qualification programs with annual competency testing
- Install full-length mirrors and step-by-step visual aids in gowning rooms
- Focus environmental monitoring rounds specifically on personnel-generated contamination zones
Equipment-Generated Contamination
Washing machines, conveyors, and inspection systems are common sources of both particle shedding and bioburden accumulation if not properly specified and maintained.
- Source cleanroom-grade equipment with smooth, non-shedding surfaces
- Establish documented cleaning and sanitization protocols for all process equipment
- Complete equipment qualification (IQ/OQ/PQ) with particle generation testing included
Maintaining Classification During Routine Operations
A cleanroom that passes qualification but fails during live production signals a design or testing gap — often because qualification didn't reflect real operational conditions.
- Design for realistic loads: actual personnel counts, equipment density, and material flow rates
- Qualify in the "operational" state with full staffing and a production simulation running
- Deploy continuous monitoring systems for real-time particle and pressure feedback
- Establish change control procedures covering all process and facility modifications
Frequently Asked Questions
What ISO cleanroom class is required for manufacturing pharmaceutical stoppers?
The required class is typically ISO 7 or ISO 8, depending on sterilization method. Stoppers for terminally sterilized products can be prepared in ISO 8 (Grade D), while aseptically filled products require ISO 7 (Grade C). Final classification is determined by risk assessment covering drug application, patient population, and target regulatory market.
How does ISO 14644 differ from GMP cleanroom classifications?
ISO 14644 is a particle-count-based international standard defining air cleanliness. GMP grades (A-D) are pharmaceutical-specific classifications that reference ISO standards but add operational requirements, microbial limits, and specific process controls. Grade C equals ISO 7 at rest but ISO 8 in operation.
What are the particle count limits for an ISO 7 cleanroom?
ISO 7 allows maximum 352,000 particles ≥0.5 microns per cubic meter and 2,930 particles ≥5.0 microns per cubic meter in operational state. At rest, limits are typically one grade better (ISO 6 equivalent).
Do rubber stoppers need to be manufactured in a cleanroom?
Elastomer compounding and molding may occur in controlled but non-classified areas. Post-molding steps — washing, depyrogenation, inspection, siliconization, and packaging — must occur in ISO 7 or ISO 8 classified cleanrooms to meet FDA and EMA requirements for components that contact sterile drugs.
What environmental monitoring is required in stopper manufacturing cleanrooms?
Required monitoring includes non-viable particle counting (continuous for Grade A, periodic for C/D), viable air and surface sampling, continuous pressure differential logging, and temperature/humidity control. ISO 14644-2 and EU GMP Annex 1 define the applicable frequencies and limits.
How often must cleanrooms be recertified for stopper production?
EU GMP Annex 1 requires recertification every 6 months for Grade A/B and every 12 months for Grade C/D. Additional testing is required after significant HVAC modifications, filter changes, or contamination events. ISO 14644-2 suggests risk-based intervals but pharmaceutical applications follow the more stringent Annex 1 requirements.


