
Introduction
Contamination drives 14% of severe drug recalls in pharmaceutical manufacturing, with microbial contamination causing an average of 330 drug recalls annually between 2012 and 2023. These numbers reflect a direct patient safety risk — not a paperwork problem. Cleanroom integrity determines whether a product reaches patients safely or gets pulled from shelves.
A laboratory cleanroom is a controlled environment designed to maintain airborne particle counts within defined limits. Unlike standard labs, cleanrooms use multi-stage HEPA filtration, controlled pressure differentials, and strict material protocols to meet ISO 14644-1 classification requirements.
These environments are critical across a range of applications — pharmaceutical compounding, biotechnology research, medical device manufacturing, and R&D — where even microscopic contamination can compromise results or product safety.
What follows covers the key elements of effective cleanroom design:
- Core design features that maintain contamination control
- ISO classification standards and what they require in practice
- HVAC and filtration systems that create the controlled environment
- Industry-specific best practices for pharmaceutical, biotech, and medical device settings
TLDR: Key Takeaways for Laboratory Cleanroom Design
- HEPA filtration, 10–15 Pa pressure differentials, and 30–50+ air changes per hour keep airborne particulates in check
- ISO Classes 5-8 define particle limits for labs: ISO 5 for sterile compounding, ISO 7 for pharmaceutical manufacturing, ISO 8 for packaging areas
- Unidirectional airflow, non-shedding surfaces, proper gowning sequences, and continuous monitoring are non-negotiable design requirements
- Modular construction reduces installation time from months to weeks while providing flexibility for future reconfiguration and expansion
- Regulatory compliance goes beyond ISO particle counts: Health Canada, FDA, and EU GMP all require microbiological limits and dynamic in-operation monitoring
Essential Design Features of Laboratory Cleanrooms
Pressure Differential Control and Contamination Prevention
Pressure cascades form the foundation of contamination control. Positive pressure rooms maintain 10-15 Pascals (0.04-0.06 inches water gauge) above adjacent spaces, preventing uncontrolled air from entering sterile production areas. Negative pressure rooms serve containment applications—protecting personnel and the environment when handling hazardous materials or pathogens.
Regulatory minimums vary by framework:
- FDA: 10-15 Pa between adjacent rooms of different classification
- EU GMP Annex 1: Minimum 10 Pa between different grades
- USP <797>: 0.02 inches water column (~5 Pa) between ISO-classified areas
- ISPE: 5 Pa practical minimum, even between rooms of the same classification

Pressure monitoring must be continuous, not periodic. FDA 483 observations frequently cite failures to monitor pressure differentials during actual production, leading to undetected contamination ingress during door openings or HVAC fluctuations.
Air Changes Per Hour Requirements
Air change rates dilute and remove contaminants, directly impacting achievable cleanliness levels:
- ISO Class 7 Buffer Rooms: 30 ACH minimum (USP <797>), though ASHRAE suggests 50 ACH for robust performance
- ISO Class 8 Ante-Rooms: 20 ACH minimum
- Hazardous Drug Areas: 12 ACH externally exhausted (ASHRAE)
Higher ACH rates demand larger HVAC systems, increasing both capital costs and ongoing energy consumption. Cleanrooms consume up to 15 times more energy than standard commercial buildings, with over 50% attributed to HVAC systems. Design must balance contamination control requirements with operational costs.
Personnel and Material Flow Design
Flow pattern design determines how effectively a cleanroom prevents cross-contamination between zones. Each transition point — from gowning rooms to airlocks to pass-throughs — should create a deliberate barrier rather than a gap.
- Airlocks buffer transitions between cleanliness levels, with interlocking doors that prevent simultaneous opening
- Gowning rooms enforce proper sequencing between street clothes and cleanroom garments
- Pass-through chambers move materials across zones without requiring personnel to cross
- Dedicated "clean" corridors serve classified rooms; separate "dirty" corridors handle waste and equipment removal
The correct gowning sequence runs from dirtiest to cleanest:
- Shoe covers
- Head and beard covers
- Face mask
- Hand hygiene
- Gown
- Sterile gloves (donned inside the classified space)
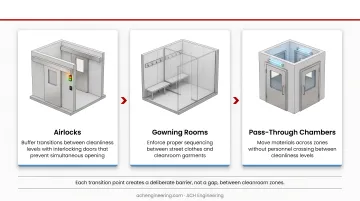
USP <797> prohibits exposed skin in buffer rooms for Category 3 compounding.
Cleanroom Layout and Airflow Optimization
Zoning separates activities by contamination risk:
- Critical processing (ISO 5) stays in dedicated zones with unidirectional airflow
- Support activities — solution preparation, equipment cleaning — occupy lower classifications
- Particle-generating equipment is isolated or enclosed with local exhaust
Maintain unobstructed airflow paths from ceiling supply to floor returns. Equipment placement must not create dead zones or redirect air from lower-grade areas toward critical zones. Airflow visualization (smoke studies) validates that air flows correctly during all operational conditions.
Monitoring and Control System Integration
Periodic testing misses transient excursions — continuous monitoring is the only reliable approach. Key parameters to track in real time:
- Particle counts in ISO 5 zones throughout production
- Pressure differentials with daily log review (USP <797> requirement)
- Temperature (20-22°C) and humidity (30-50% RH for labs; 30-60% RH general range)
- Viable air counts in ISO 5 zones — non-compliance here is a frequent FDA 483 citation
All alarms require documentation and investigation. Building automation systems should trigger alerts before conditions exceed action limits, allowing corrective intervention before reaching regulatory thresholds.
Modular vs. Traditional Construction
Modular systems offer clear advantages for most laboratory applications:
- Faster to install — weeks instead of months for equivalent traditional builds
- Reconfigurable as research needs shift, without tearing out fixed infrastructure
- Lower total cost through reduced on-site labour and material waste
- Minimal disruption to adjacent operations during installation
ACH Engineering's modular approach uses prefabricated wall panels, walkable ceilings, and flush-mount doors manufactured off-site for rapid on-site assembly. Components can be expanded, relocated, or reconfigured without major reconstruction, protecting long-term investment as laboratory needs change. Working with an experienced cleanroom consulting team early in the process ensures modular configurations are correctly specified for your ISO classification and regulatory requirements from the outset.
Traditional stick-built construction still has a place in very large facilities or projects with unusual structural demands — but for most laboratory builds, modular solutions reduce both project timelines and lifecycle costs.
ISO Classification Standards for Laboratory Cleanrooms
Understanding ISO 14644-1 Classifications
ISO 14644-1 defines cleanroom classes by maximum allowable particle concentrations per cubic meter of air. The classification follows a logarithmic scale—each class number increase represents approximately 10× more particles:
| ISO Class | 0.5 µm particles/m³ | 5.0 µm particles/m³ | Common Lab Application |
|---|---|---|---|
| ISO 5 | 3,520 | 29 | Sterile compounding, aseptic filling |
| ISO 6 | 35,200 | 293 | Cell culture, critical research |
| ISO 7 | 352,000 | 2,930 | Pharmaceutical manufacturing |
| ISO 8 | 3,520,000 | 29,300 | Packaging, equipment cleaning |
Laboratory Applications by ISO Class
ISO Class 5 (Grade A) covers the most critical applications, where product exposure risk is highest:
- Primary Engineering Controls (PECs) for sterile compounding
- Aseptic filling and product exposure zones
- Cell culture work requiring sterile technique
- Open product handling in pharmaceutical manufacturing
ISO Class 7 (Grade B/C) typically serves as the buffer environment around ISO 5 zones. Common applications include solution preparation, general pharmaceutical manufacturing, and biotechnology production areas.
ISO Class 8 (Grade D) supports lower-risk tasks:
- Ante-rooms and gowning areas
- Equipment cleaning and staging
- Packaging operations
- Material transfer zones
ISO Standards vs. GMP Requirements
ISO 14644-1 sets baseline particle limits, but GMP regulations layer on additional requirements that go further:
- FDA GMP: Adopts ISO particle limits but emphasizes microbiological data and dynamic monitoring
- EU GMP Annex 1: Defines Grades A–D with specific microbiological limits (Grade A requires zero growth for settle plates)
- USP <797>: Maps ISO classes to compounding areas with explicit air change and pressure requirements
- Health Canada GUI-0119: Applies equivalent classifications for sterile drug manufacturing facilities in Canada
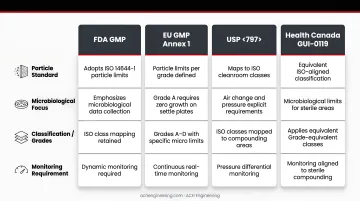
ISO standards measure particles. GMP regulations demand proof of microbiological control. A room meeting ISO 5 particle counts can still fail GMP requirements if microbiological monitoring shows contamination.
Testing and Certification Requirements
Certification intervals vary by regulatory framework:
- USP <797>: Every 6 months for ISO 5, 7, and 8 areas
- EU GMP Annex 1: Every 6 months for Grade A/B; annually for Grade C/D
- Testing conditions: Compliance must be demonstrated under dynamic (in-operation) conditions, not just at-rest
Particle counts must meet class limits at every sampling location. A single excursion triggers a mandatory investigation and corrective action before operations resume.
Determining Required ISO Classification
Conduct a contamination risk assessment based on:
- Product sensitivity: Sterile injectables demand higher classification than oral solid dosages
- Process exposure: Open product handling requires ISO 5; closed systems may allow ISO 7
- Regulatory framework: Cross-reference FDA, USP, EU GMP, or Health Canada guidance for your application
- Duration of exposure: Longer open-product exposure times typically require tighter classifications
Specific classification requirements by application:
- Pharmaceutical compounding: USP <797> mandates ISO 5 PECs within ISO 7 buffer rooms
- Medical devices: Reference FDA 21 CFR Part 820 alongside your device risk classification
- Research laboratories: ISO 6–7 for cell culture; ISO 7–8 for general laboratory work
HVAC and Air Filtration Systems
HEPA vs. ULPA Filter Efficiency
Two filter standards dominate cleanroom design, each suited to different ISO classifications:
- HEPA (High-Efficiency Particulate Air) filters capture at least 99.97% of particles at 0.3 µm — the hardest size to trap. They meet requirements for ISO Classes 5–8 and are tested to IEST-RP-CC001 (North America) or ISO 29463 (global).
- ULPA (Ultra-Low Penetration Air) filters reach 99.999% efficiency at the Most Penetrating Particle Size (MPPS), reserved for ISO Classes 1–4 or demanding semiconductor and nanotechnology applications.
Once installed, filters must be verified. Integrity testing uses an aerosol challenge (PAO or DOP) with photometric scanning of the downstream filter face — any probe reading at 0.01% of the upstream challenge signals a leak requiring repair or replacement. FDA and Health Canada guidance for aseptic processing rooms both recommend testing at least twice annually.
Laminar vs. Turbulent Airflow
The filter type selected feeds directly into the airflow strategy — and choosing the wrong approach for your ISO classification wastes both energy and capital.
Unidirectional (laminar) airflow moves air at uniform velocity across the entire work zone, physically sweeping particles away from critical surfaces rather than simply diluting them. EU GMP Annex 1 specifies 0.36–0.54 m/s homogeneous air speed for Grade A zones, measured 150–300 mm from the filter face.
Typical applications: ISO Class 5 critical zones, biological safety cabinets, aseptic filling lines.
Turbulent (non-unidirectional) airflow uses high-volume mixing ventilation to dilute contaminants. It costs less to install than unidirectional systems but requires higher air change rates to achieve comparable cleanliness levels.
Typical applications: ISO Classes 6–8, buffer rooms, ante-rooms, general laboratory spaces.
Fan Filter Units (FFUs) integrate a HEPA filter and fan into a single ceiling-mounted module, delivering uniform airflow without complex supply ductwork. They eliminate the need for a centralized air handling system, making them a natural fit for modular cleanroom builds where layout flexibility matters.
HVAC System Design Considerations
Complete system architecture includes:
- Outdoor air handlers: Condition makeup air to temperature/humidity setpoints
- Pre-filtration stages: MERV 8-14 filters protect final HEPA filters from premature loading
- Temperature control: 18-22°C typical range; USP <797> requires ≤20°C for compounding areas
- Humidity control: 30-60% RH prevents microbial growth and static electricity; USP <797> requires ≤60% RH
- Energy recovery: Heat wheels or plate exchangers reduce conditioning costs for exhaust air
Energy efficiency strategies:
- Variable frequency drives (VFDs) on supply fans to modulate airflow based on occupancy
- Demand-controlled ventilation reducing ACH during unoccupied periods (where regulations permit)
- LED lighting reducing heat load and HVAC demand
- Optimized pressure differentials (higher differentials increase energy consumption)
Best Practices for Cleanroom Construction and Layout
Optimal Pressurization Strategies
Recommended pressure differentials:
- 10-15 Pa (0.04-0.06" w.g.) between adjacent rooms of different classification (FDA guidance)
- Minimum 5 Pa for robust control even between same-class rooms (ISPE recommendation)
- Continuous monitoring with daily log review and automated alarms
Pressure cascade design: ISO 5 → ISO 7 → ISO 8 → unclassified space, with each transition maintaining minimum differential. Airlocks create intermediate zones, allowing doors to open without direct connection between classification extremes.
Common failure point: Inadequate door seals or simultaneous opening of interlocked doors causes pressure reversals. Design must account for worst-case scenarios during normal operations.
Minimizing Particle Generation Sources
Strategic equipment placement:
- Isolate particle-generating equipment (centrifuges, lyophilizers) in separate rooms or enclosures
- Provide local exhaust for equipment producing heat, moisture, or particles
- Use vibration isolation for machinery to prevent particle resuspension
Material selection:
- Stainless steel or powder-coated surfaces (not particle-shedding materials)
- Sealed motors and drives preventing internal particle escape
- Cleanroom-rated casters and wheels on mobile equipment
Gowning and De-Gowning Protocols
Proper anteroom design includes:
- Separate gowning and de-gowning areas to prevent cross-contamination
- Hand washing stations with hands-free controls
- Sticky mats at entry points capturing shoe-borne particles
- Adequate space for multiple personnel (minimum 8-10 sq ft per person)
- Mirrors for self-inspection of garment donning
Gowning sequence (clean to cleanest):
- Shoe covers
- Head/beard covers
- Face mask
- Hand hygiene (wash or sanitize)
- Cleanroom gown
- Sterile gloves (in classified area)
Personnel remain the largest contamination source. EU GMP Annex 1 requires documented validation of aseptic gowning practices, and regular competency assessments keep personnel compliant between audits.
Cleaning and Maintenance Protocols
Effective gowning reduces what personnel bring in — cleaning protocols address what accumulates on surfaces regardless. Frequency varies by classification:
- ISO 5: Daily cleaning of all surfaces
- ISO 7: Daily cleaning of floors; weekly cleaning of walls/ceilings
- ISO 8: Weekly cleaning of all surfaces
Appropriate cleaning agents:
- Sterile 70% isopropyl alcohol for disinfection
- Sporicidal agents (hydrogen peroxide, peracetic acid) for periodic deep cleaning
- Rotation of disinfectants to prevent resistant organism development
Cleaning validation must demonstrate reduction of residues to validated limits, with documented effectiveness against facility-specific microorganisms as required by EU GMP Annex 1.
Validation and Qualification Processes
Once cleaning protocols are established, the facility moves into formal qualification — the structured process that proves the cleanroom performs as designed before production begins.
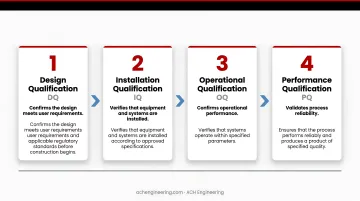
Four-phase qualification approach:
- Design Qualification (DQ): Confirms the design meets user requirements and applicable regulatory standards before construction begins
- Installation Qualification (IQ): Verifies that equipment and systems are installed according to approved specifications
- Operational Qualification (OQ): Tests that all systems perform within specified ranges across the full operating envelope
- Performance Qualification (PQ): Runs the cleanroom under real production conditions to confirm sustained, repeatable results
Documentation requirements:
- Detailed protocols specifying acceptance criteria
- Test results with raw data and calculations
- Deviation reports and corrective actions
- Final summary report with approval signatures
Work with accredited certification bodies familiar with your industry's regulatory requirements. ACH Engineering's cleanroom validation services support clients through each qualification phase, coordinating test protocols and documentation to meet certification requirements before handover.
Future-Proofing Cleanroom Designs
Design for scalability:
- Modular construction allowing easy expansion without major reconstruction
- Oversized HVAC systems accommodating additional zones
- Flexible utility distribution (electrical, gases, process water)
- Strategic placement of mechanical rooms for future connections
Technology accommodation:
- Adequate power capacity for evolving equipment
- Structured cabling for monitoring systems
- Ceiling space for additional FFUs or lighting
- Floor loading capacity for heavy equipment
Regulatory adaptability:
- Design margins exceeding current requirements
- Monitoring systems capable of tighter specifications
- Documentation systems supporting future audits
ACH Engineering's modular construction approach makes this adaptability practical: additional airlocks, gowning rooms, or entire cleanroom sections can be integrated into existing installations without major reconstruction as operational needs change.
Material Selection and Surface Requirements
Wall, Ceiling, and Floor Materials
Essential properties for cleanroom surfaces:
- Non-shedding (no particle generation)
- Smooth and impervious (easy cleaning)
- Chemical resistant (withstand disinfectants)
- Seamless or sealed joints (no particle accumulation)
Preferred materials:
- Walls: Powder-coated steel panels, High-Pressure Laminate (HPL), or u-PVC modular panels
- Ceilings: Sealed lay-in panels or walk-on grid systems with smooth lower surfaces
- Floors: Heat-welded vinyl, epoxy coatings, or seamless sheet flooring
Coved transitions between walls and floors/ceilings eliminate 90-degree corners where particles accumulate and cleaning is difficult. EU GMP Annex 1 and cGMP regulations require these seamless connections. ACH Engineering's cleanroom fabrication — wall panel systems available in HPL, powder-coated galvanized iron, and u-PVC — are prefabricated and modular, supporting fast installation while meeting ISO and cGMP standards for surface continuity.
Lighting Requirements
Illumination levels:
- General cleanrooms: 800–1,000 lux (75–93 foot-candles)
- Detailed inspection work: 1,500–2,000 lux
- Photolithography: Yellow lighting (sodium vapor) blocking UV wavelengths
Fixture requirements:
- Sealed, flush-mounted designs preventing particle accumulation
- Cleanroom-rated gaskets and lenses
- LED technology reducing heat load and energy consumption
- Chemical resistance to cleaning agents
Avoid recessed fixtures with ledges or exposed fasteners. All penetrations through ceiling or wall surfaces must be sealed.
Doors, Windows, and Pass-Throughs
Each opening type has distinct requirements that affect both contamination control and pressure management:
- Doors: Flush surfaces without handles, ledges, or exposed hinges; interlocking mechanisms on airlocks
- Windows: Flush-mounted with sealed frames; no external ledges
- Pass-throughs: Interlocked doors preventing simultaneous opening; smooth interior surfaces; UV sterilization options
- Frames: Powder-coated steel or stainless steel with tempered glass or acrylic vision panels
- Gaskets: Sized to maintain pressure differentials and block particle infiltration
Avoid sliding doors in critical zones — exposed tracks are hard to clean and rarely maintain reliable seals.
Industry-Specific Laboratory Cleanroom Requirements
Pharmaceutical and Compounding Pharmacy Requirements
USP <797> sterile compounding mandates:
- ISO Class 5 PEC (Primary Engineering Control): Laminar airflow hood or isolator
- ISO Class 7 buffer room: Surrounds PEC; minimum 30 ACH
- ISO Class 8 ante-room: Positive pressure buffer; minimum 20 ACH
- Certification: Every 6 months under dynamic conditions
- Temperature: ≤20°C (68°F)
- Humidity: ≤60% RH
USP <800> hazardous drug handling requires:
- Containment Secondary Engineering Control (C-SEC): Negative pressure relative to surrounding areas
- External venting: Minimum 12 ACH exhausted outside
- No recirculation: All air from hazardous areas must be exhausted
Category 3 compounding (high-risk preparations) prohibits exposed skin (face/neck) in buffer rooms, requiring full coverage with sterile garments.
Biotech and Research Laboratory Needs
Biosafety Level (BSL) considerations:
- BSL-1/2: Standard microbiological practices; may not require classified cleanrooms
- BSL-3: Negative pressure containment; HEPA-filtered exhaust; sealed penetrations
- BSL-4: Maximum containment; Class III biological safety cabinets; airlocks with shower decontamination
BSL classification focuses on biological containment — protecting personnel and the environment — while ISO classification targets particulate cleanliness to protect the product. A facility can be both BSL-3 and ISO Class 7, but the pressure requirements directly conflict: BSL-3 demands negative pressure for containment, while ISO Class 7 typically uses positive pressure for particle control. Engineering both simultaneously requires careful zoning and airflow analysis — a challenge best addressed through purpose-built controlled environment systems for research facilities.

Specialized equipment integration:
- Cell culture incubators requiring temperature/CO₂ control
- Ultra-low temperature freezers generating considerable heat load
- Genomics equipment (sequencers, PCR machines) needing vibration isolation
- Microbiology equipment (autoclaves, incubators) requiring dedicated utilities
Medical Device Manufacturing Requirements
FDA 21 CFR Part 820 requires environmental controls where conditions could adversely affect product quality. Classification depends on device risk:
- Class III devices (high risk): Typically require ISO 5-7 for sterile manufacturing
- Class II devices (moderate risk): ISO 7-8 for sterile products; may not require classified environment for non-sterile
- Class I devices (low risk): Generally no cleanroom requirement unless sterile
Whether a device requires sterile or non-sterile production significantly shapes the environmental design:
- Sterile devices demand validated aseptic processing or terminal sterilization
- Non-sterile devices may require only controlled (not classified) environments
- Risk assessment determines appropriate classification based on patient contact and infection potential
Common Mistakes and Challenges in Cleanroom Design
Frequent Design Errors
- Specifying minimum 30 ACH for ISO 7 without accounting for equipment heat load — high-density areas may need 50+ ACH to hold temperature and particle counts
- Designing for 5 Pa pressure differential without factoring in door openings, which can trigger temporary reversals; 10-15 Pa is the safer target for all operational scenarios
- Undersizing gowning areas — crowded spaces increase contamination risk; plan for 8-10 sq ft per person with separate entry and exit paths
- Undersizing HVAC capacity because initial design excludes future equipment; build in a 20-30% margin from the start
- Skipping smoke studies and relying on calculated airflow patterns alone — physical testing routinely uncovers unexpected air currents from diffuser placement, return locations, or equipment position

Even a well-designed cleanroom will underperform if operational practices don't keep pace. These are the challenges facilities encounter most often after commissioning.
Common Operational Challenges
- Even a well-designed cleanroom fails without consistent gowning protocols, controlled movement, and scheduled cleaning — ongoing staff training and periodic assessments are non-negotiable
- Cost-cutting pressure can push teams toward ACH reductions or delayed filter replacements; energy optimization is valid, but regulatory compliance cannot be traded for cost savings
- Semi-annual certification under USP <797> carries real ongoing cost — particle counting, airflow testing, and microbiological monitoring should be budgeted as recurring operational expenses, not one-time capital items
- Regulatory agencies increasingly scrutinize continuous monitoring records; automated data logging reduces the compliance burden, though it requires upfront investment
Frequently Asked Questions
What is a lab cleanroom?
A lab cleanroom is a controlled environment designed to maintain low levels of airborne particles through HEPA filtration, pressure differentials, and strict entry protocols. Applications span pharmaceutical compounding, biotechnology research, medical device manufacturing, and R&D settings where contamination could compromise results or product safety.
What are the different types of cleanrooms?
Cleanrooms differ across three main dimensions:
- Construction: Hardwall prefabricated panels vs. softwall vinyl curtains
- Pressure control: Positive pressure for sterile production vs. negative pressure for hazardous containment
- ISO classification: Class 5–8 covers most laboratory applications
Your specific process, budget, and regulatory requirements determine which combination fits best.
What ISO class cleanroom do I need for my laboratory?
ISO 5 suits sterile compounding and aseptic processes with open product exposure. ISO 6–7 covers general pharmaceutical manufacturing and cell culture. ISO 8 works for packaging and support areas. A contamination risk assessment — weighing product sensitivity, process exposure, and regulatory obligations — determines the right classification.
How much does it cost to build a laboratory cleanroom?
Costs range from $200–$550 per square foot (CAD) depending on ISO classification, size, and construction type. ISO 8 modular cleanrooms typically run $200–$350/sq ft; ISO 7 ranges $250–$500/sq ft. Modular construction generally reduces costs through lower labor requirements and faster installation compared to traditional builds.
What are the key differences between modular and traditional cleanroom construction?
Modular cleanrooms install in weeks vs. months for traditional construction, using prefabricated components manufactured off-site for rapid on-site assembly. They offer easier future reconfiguration, reduced installation disruption, and often lower total costs. Traditional construction may suit very large facilities or unique architectural requirements but lacks modular flexibility.
How do you maintain proper airflow in a laboratory cleanroom?
Four practices keep airflow performing as designed:
- Replace HEPA filters annually or when pressure drop rises 20%
- Monitor pressure differentials continuously, reviewing logs daily
- Keep ceiling supply-to-floor return paths clear of obstructions
- Run airflow visualization and particle count tests every 6 months per USP <797>
Building automation systems simplify continuous monitoring and alert staff to deviations before they become compliance issues.


