
Introduction
Cleanroom failures cost lives and hundreds of millions in product recalls — the FDA reported over $900 million in pharmaceutical recall costs in a single recent year, with contamination among the leading causes. A single contamination event can compromise entire production batches, trigger regulatory action, and jeopardize patient safety. For companies operating in pharmaceuticals, medical devices, and biotechnology, ISO 14644 compliance underpins every quality assurance programme and regulatory approval pathway.
ISO 14644 standards define the international framework for cleanroom classification and controlled environments, covering particle count limits, monitoring protocols, personnel requirements, and environmental controls. Recent 2024–2025 updates have introduced stricter documentation requirements, risk-based operational approaches, enhanced contamination control protocols, and new energy efficiency considerations — each with direct implications for how facilities design, certify, and operate.
This guide covers current ISO 14644 standards, recent revisions including the significant ISO 14644-5:2025 update, compliance requirements across all operational states, and practical steps for achieving certification while adapting to evolving regulatory expectations.
TLDR:
- ISO 14644 defines 9 cleanroom classes (Class 1–9) based on particle concentration limits
- ISO 14644-5:2025 now mandates formal Operations Control Programmes with risk-based protocols
- Compliance requires testing across three states: as-built, at-rest, and operational
- ISO Class 7 aligns with EU GMP Grade C at-rest but differs during operation
- Recent updates emphasize documentation, energy efficiency, and contamination control
Understanding ISO 14644 Standards: A Comprehensive Overview
What ISO 14644 Is and Why It Matters
For any facility where contamination directly affects product quality or patient safety, ISO 14644 defines the rules of the game. Developed by ISO Technical Committee 209, it is the international standard for cleanroom classification and controlled environments — covering design, construction, operation, testing, and monitoring requirements.
Key Parts of ISO 14644:
- Part 1 (ISO 14644-1:2015) — classifies air cleanliness by particle concentration; the foundation for all cleanroom grades
- Part 2 (ISO 14644-2:2015) — sets monitoring requirements to provide ongoing evidence of cleanroom performance
- Part 3 (ISO 14644-3:2019) — defines test methods for airborne particles, airflow, pressure differentials, and recovery
- Part 4 (ISO 14644-4:2001) — covers design, construction, and start-up requirements
- Part 5 (ISO 14644-5:2025) — updated in 2025 with mandatory Operations Control Programmes
- Part 7 — governs separative devices including clean air hoods, gloveboxes, and isolators
- Part 8 — classifies air cleanliness by chemical concentration
- Part 9 — classifies surface cleanliness by particle concentration

The ISO Classification System: Class 1 Through 9
Those parts translate into a nine-class hierarchy. ISO 14644-1:2015 defines each class by the maximum allowable particle concentrations per cubic meter, measured at specific particle sizes in micrometers (µm).
ISO Cleanroom Classifications:
| ISO Class | Particles ≥0.1 µm/m³ | Particles ≥0.5 µm/m³ | Particles ≥1.0 µm/m³ | Particles ≥5.0 µm/m³ |
|---|---|---|---|---|
| Class 1 | 10 | — | — | — |
| Class 2 | 100 | 4 | — | — |
| Class 3 | 1,000 | 35 | 8 | — |
| Class 4 | 10,000 | 352 | 83 | — |
| Class 5 | 100,000 | 3,520 | 832 | — |
| Class 6 | 1,000,000 | 35,200 | 8,320 | 293 |
| Class 7 | — | 352,000 | 83,200 | 2,930 |
| Class 8 | — | 3,520,000 | 832,000 | 29,300 |
| Class 9 | — | 35,200,000 | 8,320,000 | 293,000 |
ISO Class 1 represents the most stringent classification, permitting only 10 particles ≥0.1 µm per cubic meter—reserved for nanotechnology and the most critical semiconductor manufacturing processes.
Industries That Rely on ISO 14644 Compliance
ISO 14644 compliance is non-negotiable across several regulated sectors:
- Pharmaceutical and biotech manufacturers use Classes 5–7 for sterile drug production, aseptic processing, and biologics to prevent microbial and particulate contamination
- Medical device producers — from surgical instruments to implants — require controlled environments to meet sterility requirements
- Semiconductor fabs operate at Classes 1–4, where a single nanoscale particle can cause catastrophic yield loss
- Aerospace facilities control contamination during component assembly and testing to protect flight safety
- Food and beverage processors rely on ISO-compliant environments for aseptic packaging and sensitive production lines
How ISO 14644 Relates to Other Standards
Understanding which ISO class your facility needs is only part of the compliance picture. Regulatory bodies layer their own requirements on top of the ISO baseline:
EU GMP Annex 1 (2022) Alignment:
EU GMP Grade classifications map to ISO classes but with operational distinctions:
- Grade A: ISO Class 5 at-rest and in-operation
- Grade B: ISO Class 5 at-rest, ISO Class 7 in-operation
- Grade C: ISO Class 7 at-rest, ISO Class 8 in-operation
- Grade D: ISO Class 8 at-rest
FDA Requirements:
The FDA references ISO 14644 in guidance documents but adds specific requirements for sterile drug manufacturing, including airflow velocity recommendations of 0.45 m/s ± 20% for unidirectional flow systems.
Recent Updates to ISO 14644 Standards in 2024-2025
ISO 14644-5:2025 – Major Operational Overhaul
The 2025 revision of ISO 14644-5 represents the most significant update to cleanroom standards in over two decades. Where previous editions offered general guidance, this revision sets specific, enforceable operational requirements that facilities must meet.
Operations Control Programme (OCP)
Facilities must now establish a formal, documented OCP governing all operational aspects. The OCP must integrate:
- Risk assessment methodologies for identifying contamination sources
- Detailed cleaning protocols with scientific justification for frequencies
- Personnel training programs with competency verification
- Material flow procedures to minimize contamination introduction
- Gowning protocols appropriate to each cleanroom classification
Risk-Based Operational Approaches
ISO 14644-5:2025 requires facilities to apply risk assessment throughout their operational protocols — moving away from prescriptive, one-size-fits-all rules. In practice, this means:
- Justify sampling locations using airflow visualization studies
- Establish monitoring frequencies based on historical performance data
- Document rationale for cleaning procedures rather than following arbitrary schedules
- Implement change control that evaluates contamination risk before modifications
Enhanced Documentation and Record-Keeping
Documentation requirements have been substantially tightened. Regulators and auditors will now expect evidence-based records at every stage — not just end-of-project sign-offs. Specifically:
- Design Documentation: Must include contamination control strategy, material selection justification, and airflow modeling results
- Qualification Records: IQ/OQ/PQ protocols must demonstrate compliance with classification requirements across all operational states
- Ongoing Monitoring: Continuous environmental monitoring data must be trended with statistical analysis to detect adverse patterns before excursions occur
- Deviation Management: Any classification excursion requires documented investigation, root cause analysis, and corrective action verification
Stricter Contamination Control Protocols
Surface-level contamination control has received sharper focus in the updated standards. Facilities relying on legacy monitoring methods will need to reassess their approach against new requirements that include:
- Enhanced requirements for surface monitoring using ISO 14644-9 methodologies
- Specific guidance on preventing cross-contamination during material transfer
- Stricter personnel behavior protocols, particularly regarding unidirectional airflow disruption
- Integration of consumables qualification requirements from ISO 14644-18 (2023)
Energy Efficiency and Sustainability Guidance
Beyond contamination control, 2024-2025 has renewed attention on ISO 14644-16:2019 as facilities look to balance sustainability goals with compliance obligations. Key approaches gaining traction include:
- Variable air volume (VAV) systems that adjust air change rates based on real-time particle monitoring
- Risk-based reductions in air change rates during unoccupied periods
- Case studies demonstrate 14% energy reductions without compromising classification
Updated Testing and Monitoring Frequencies
The 2025 revision also brings clearer guidance on re-certification timelines — an area that has historically been inconsistent across facilities. Updated requirements include:
- Particle count testing frequency based on cleanroom class and criticality
- Recovery testing required after significant HVAC modifications
- Continuous monitoring systems must include alarm limits based on statistical analysis of historical data
- Annual re-certification recommended for most pharmaceutical applications, with risk assessment justifying extended intervals
Key Compliance Requirements Under ISO 14644
Three Operational States for Classification
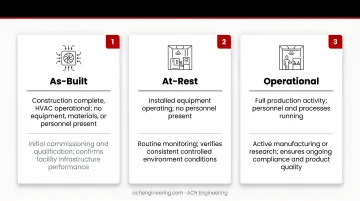
ISO 14644-1 defines three distinct occupancy states that determine when and how classification testing occurs:
| State | Conditions | Primary Use |
|---|---|---|
| As-Built | Construction complete, HVAC operational; no equipment, materials, or personnel present | Initial commissioning and qualification; confirms facility infrastructure meets design specs |
| At-Rest | Equipment installed and operating; no personnel present | Baseline performance testing; EU GMP Grade classifications align with ISO classes in this state |
| Operational | Normal production with specified personnel, equipment running, materials present | Real-world contamination control assessment; most demanding state due to process-generated particles |
Required Performance Tests
ISO 14644-3:2019 specifies the test methods required to demonstrate compliance:
Particle Count Test — Primary classification test using a Light Scattering Airborne Particle Counter (LSAPC), calibrated per ISO 21501-4. Sample volume must be sufficient to detect a minimum of 20 particles if concentration were at the class limit; at least 2 liters sampled over a minimum of 1 minute per location.
Airflow Velocity/Volume Test — Measures unidirectional flow velocity or air change rates for non-unidirectional systems. EU GMP Annex 1 specifies 0.36–0.54 m/s at the working position; FDA guidance recommends 0.45 m/s ± 20%. Confirms HVAC delivers designed air volumes.
Pressure Differential Test — Confirms positive pressure between adjacent cleanroom areas. EU GMP requires a minimum 10–15 Pascals (Pa) between grades, maintained continuously during operation to prevent contamination migration from lower-classified zones.
Recovery Test — Measures the time required to return to specified cleanliness after particle injection. Particularly critical for non-unidirectional airflow systems, and required after significant HVAC modifications.
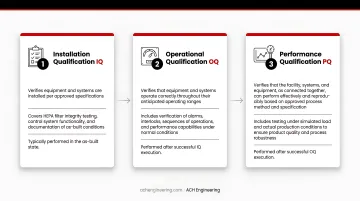
Qualification Process: IQ, OQ, and PQ
Once performance tests confirm your system meets design parameters, formal qualification documents that evidence for regulators and auditors. The process runs in three stages:
Installation Qualification (IQ) — Verifies equipment and systems are installed per approved specifications. Covers HEPA filter integrity testing, control system functionality, and documentation of as-built conditions. Typically performed in the as-built state.
Operational Qualification (OQ) — Confirms the system operates across its full anticipated range. Includes airflow testing, pressure differential verification, and temperature/humidity mapping — usually at-rest to isolate system performance from personnel variables.
Performance Qualification (PQ) — Demonstrates the system performs under routine operating conditions, with particle count testing in the operational state and personnel present. Includes worst-case scenarios such as maximum occupancy to confirm contamination control holds under real production load.
Ongoing Monitoring Requirements
Qualification establishes the baseline — ongoing monitoring maintains it.
Continuous Particle Monitoring:
- Real-time monitoring required in critical areas (Grade A/ISO Class 5)
- Data trending detects adverse patterns before formal excursions occur
- Alert limits typically set at the 95th percentile of historical performance data
Environmental Parameter Tracking:
- Temperature, humidity, and pressure differentials monitored continuously
- Automated data collection with data integrity controls
- Deviation investigations required when parameters exceed established ranges
Periodic Re-Certification:
- Particle count testing intervals determined by risk assessment
- Annual re-certification is standard for pharmaceutical applications
- HEPA filter integrity testing after filter changes or at minimum annually
- Airflow testing triggered by any system modifications
Documentation Requirements
Every qualification activity and monitoring event generates records that form your compliance trail:
- Standard Operating Procedures (SOPs): Cleaning, gowning, material transfer, monitoring, and deviation management
- Test Records: All qualification and monitoring results, including raw data
- Deviation Reports: Investigation records for any classification excursions
- Change Control: Impact assessments for facility or process modifications
- Training Records: Personnel qualification and ongoing competency verification
Classification and Testing Requirements
Determining Appropriate ISO Class
Selecting the correct ISO class requires evaluating:
- Product risk: Sterile injectables require stricter control (Class 5–7) than oral solid dosages (Class 7–8)
- Process requirements: Open aseptic processing demands higher classification than closed systems
- Regulatory expectations: FDA and EMA guidance specify classifications for particular operations
- Contamination sensitivity: Semiconductors require Classes 1–4 due to nanoscale sensitivities
Common Industry Classifications
- Sterile drug filling (aseptic processing): ISO Class 5 (Grade A)
- Background areas for aseptic processing: ISO Class 7 (Grade B/C)
- Non-sterile pharmaceutical manufacturing: ISO Class 7–8
- Medical device assembly: ISO Class 6–8, depending on device criticality
- Semiconductor fabrication: ISO Class 1–4
Particle Counting Methodology
Once the target ISO class is set, particle counting confirms whether the environment actually meets it. Accurate measurement requires following ISO 14644-1:2015 sampling protocols:
Sampling Location Calculation
For cleanrooms ≤1,000 m²:
- Refer to Table A.1 in ISO 14644-1
- Example: 10 m² room requires 5 sampling locations
- Example: 100 m² room requires 16 sampling locations
For cleanrooms >1,000 m²:
- Use Formula: NL = 27 × (Area/1000)
- Where NL = number of locations, Area = cleanroom area in m²
Sample Volume Requirements
- Minimum 2 liters per location — enough to statistically detect at least 20 particles at the class limit
- Sampling time minimum 1 minute per location
Location Selection
Placement matters as much as sample volume. Poorly chosen locations can mask real contamination risks:
- Divide the cleanroom into equal grid sections matching the required location count
- Select a representative point within each grid section
- Prioritize critical areas near product exposure zones
- Document location rationale using airflow studies

Statistical Analysis
- Each location must independently meet class limits (no UCL calculation required for 2–9 locations)
- Zero particle counts are valid data points indicating compliance
- Tracking counts across multiple test cycles reveals gradual filter degradation or airflow imbalances — enabling corrective action before a formal classification failure
Documentation and Monitoring Best Practices
Cleanroom Qualification Protocol Requirements
A comprehensive qualification protocol must include:
- Specific cleanroom areas covered and classification targets (scope and objectives)
- Particle count limits, airflow parameters, pressure differentials, and environmental ranges (acceptance criteria)
- Detailed test procedures referencing ISO 14644-3 methodologies
- Location maps with scientific justification for sampling point placement
- Clear assignment of execution, review, and approval responsibilities
- Documented procedures for managing out-of-specification results
Environmental Monitoring Best Practices
Effective EM programs require data-driven approaches:
Alert and Action Limits:
- Alert levels set at the 95th percentile of historical data to provide early warning
- Action limits tied to regulatory thresholds (such as Annex 1 specifications) that require immediate investigation
- Limits must be statistically justified, not arbitrary
Monitoring Parameters:
- Viable and non-viable particulates in critical areas
- Temperature and relative humidity
- Pressure differentials between adjacent areas
- Airflow velocity in unidirectional flow zones
- HEPA filter integrity (annual or post-change)
Data Trending:
- Statistical analysis to identify adverse trends before excursions
- Monthly and quarterly review of monitoring data
- Investigation triggered by upward trends even below action limits
- FDA 483s frequently cite failure to detect trends before violations occur
Change Control System
A well-structured change control system ensures modifications don't compromise cleanroom classification:
Impact Assessment:
- Evaluate potential effect on airflow patterns, particle generation, and contamination risk
- Determine if re-qualification testing is required
- Document rationale for risk determination
Change Categories:
- Minor changes require documentation but not re-testing — such as replacing like-for-like consumables
- Major changes trigger partial or full re-qualification — for example, HEPA filter replacement or HVAC modifications
Implementation:
- Controlled execution with verification testing
- Training on new procedures or equipment
- Documentation of pre- and post-change conditions
Achieving and Maintaining Compliance
Step-by-Step Compliance Process
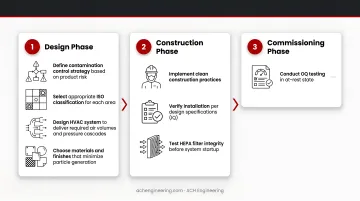
1. Design Phase:
- Define contamination control strategy based on product risk
- Select appropriate ISO classification for each area
- Design HVAC system to deliver required air volumes and pressure cascades
- Choose materials and finishes that minimize particle generation
2. Construction Phase:
- Implement clean construction practices
- Verify installation per design specifications (IQ)
- Test HEPA filter integrity before system startup
3. Commissioning Phase:
- Conduct OQ testing in at-rest state
- Verify airflow patterns, particle counts, and environmental parameters
- Establish baseline performance data
4. Operational Qualification:
- Perform PQ testing under routine operating conditions
- Include worst-case scenarios (maximum occupancy, equipment operation)
- Demonstrate classification compliance in operational state
5. Ongoing Maintenance:
- Implement continuous monitoring programs
- Conduct periodic re-certification testing
- Maintain robust documentation and change control
- Trend data to detect deterioration early
Common Compliance Challenges
Managing Contamination Events:
- Excursions require immediate investigation and corrective action
- Root cause analysis must identify systemic issues, not just symptoms
- Preventive actions should address underlying contamination sources
Maintaining Documentation:
- Electronic systems must comply with 21 CFR Part 11 for data integrity
- Paper records require secure storage and controlled access
- Training records must demonstrate ongoing competency
Balancing Energy Efficiency with Performance:
- Reducing air change rates saves energy but risks classification failure
- ISO 14644-16 guidance enables risk-based optimization
- Real-time monitoring data justifies ACR reductions without compromising cleanliness
Adapting to Standard Updates:
- ISO 14644-5:2025 requires formal Operations Control Programmes
- Facilities must update procedures and documentation to align with new requirements
- Gap assessments identify areas needing remediation
Working with Experienced Cleanroom Partners
Achieving ISO 14644 compliance requires specialized expertise in controlled environment design and construction. ACH Engineering designs and builds ISO Class 1–9 cleanrooms for pharmaceutical, biotech, and medical device clients, as well as semiconductor, and aerospace facilities across North America.
ACH Engineering's modular cleanroom solutions cover the full project lifecycle — design engineering, component manufacturing, on-site construction, and validation support — with built-in flexibility for future expansion or reconfiguration.
Frequently Asked Questions
Which ISO cleanroom is the cleanest?
ISO Class 1 is the cleanest classification, permitting only 10 particles ≥0.1 µm per cubic meter. Class 1 cleanrooms are rare and used exclusively for the most critical applications like advanced semiconductor manufacturing and nanotechnology research where even minute contamination causes catastrophic failure.
Is ISO 7 equivalent to Grade C?
ISO Class 7 aligns with EU GMP Grade C for at-rest conditions (352,000 particles/m³ ≥0.5 µm). Grade C in-operation permits 3,520,000 particles/m³, which corresponds to ISO Class 8. Facilities must meet Grade C at-rest conditions but are permitted higher particle counts during active production.
What are the new changes in ISO 14644-1?
ISO 14644-1:2015 introduced table-based sampling location determination and eliminated UCL calculations for small sample sets. The most recent update, ISO 14644-5:2025, mandates Operations Control Programmes, risk-based operational protocols, enhanced documentation, and greater energy efficiency alongside contamination control.


