
Introduction
Some facilities have recorded over 50 unreported microbial excursions in critical cleanroom zones — each one a potential patient safety failure and a regulatory citation waiting to happen. Environmental monitoring (EM) is the system designed to catch these events before they become crises, verifying that air quality, surfaces, and environmental conditions stay within validated limits throughout manufacturing.
Effective implementation isn't uniform. It varies by cleanroom classification, industry, monitoring technology, sampling strategy, and the regulatory frameworks in play — ISO 14644, EU GMP Annex 1, and FDA cGMP chief among them.
Regulatory expectations have also shifted. The 2022 EU GMP Annex 1 revision now mandates continuous monitoring for Grade A zones — periodic-only sampling during critical operations is no longer acceptable. FDA enforcement data consistently flags inadequate monitoring and personnel behavior as leading causes of warning letters.
This guide covers the full scope: when EM is required, which parameters to track, how to build and execute a compliant program, which technologies to consider, and where most facilities go wrong.
Key Takeaways
- Environmental monitoring tracks viable particles, non-viable particles, temperature, humidity, pressure differentials, and airflow to prevent cleanroom contamination
- Effective programs combine risk-based planning, calibrated equipment, and sampling locations mapped to ISO/GMP classification
- Implementation follows risk assessment → equipment setup → IQ/OQ/PQ validation → ongoing data trending and CAPA response
- Success depends on trained personnel, ALCOA+ documentation, real-time alerts, and full QMS integration
How to Implement Environmental Monitoring in Cleanroom Manufacturing
Step 1: Conduct Risk Assessment and Develop Monitoring Plan
Start by identifying critical control points based on your cleanroom classification (ISO 5-8 / EU GMP Grades A-D), process type (aseptic filling, sterile compounding, etc.), and product risk to patient safety. The 2022 EU GMP Annex 1 requires all EM programs to be built around a documented Contamination Control Strategy (CCS) covering facility design, equipment, and operations.
Map Monitoring Zones and Sampling Locations
Create a detailed cleanroom layout identifying sampling locations for air, surfaces, and personnel. Focus on areas with the highest contamination risk:
- Proximity to exposed product or open containers
- High-traffic zones where personnel movement is frequent
- Areas near equipment that may shed particles
- Points where materials enter the cleanroom
- Locations identified through smoke studies showing airflow patterns
Establish Monitoring Frequencies
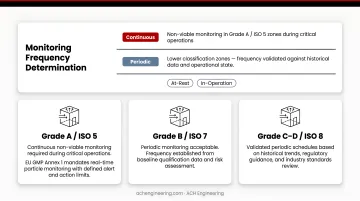
Determine whether monitoring will be continuous or periodic for each parameter and location. Current regulations require continuous non-viable monitoring in Grade A/ISO 5 zones during critical operations. Lower classifications may use validated periodic frequencies based on historical data and operational state (at-rest vs. in-operation).
Review regulatory guidance, industry standards, and baseline qualification data to establish scientifically justified limits. For Grade A zones, EU GMP Annex 1 enforces a "no growth" expectation (0 CFU) for viable monitoring — any microbial recovery requires immediate investigation. Document the rationale for all limits.
Consolidate all parameters, locations, frequencies, methods, equipment specifications, acceptance criteria, and responsibilities into a single controlled document. Obtain Quality Assurance approval before implementation begins.
Step 2: Select and Install Monitoring Equipment and Infrastructure
Research Monitoring Technologies
Compare options across these categories:
- Particle counters: Portable vs. fixed systems (continuous monitoring required for Grade A)
- Viable samplers: Active air samplers vs. passive settle plates
- Surface monitoring: Contact plates vs. swabs
- Data systems: Manual logbooks vs. automated Building Management Systems (BMS/EMS)
Evaluate Equipment Against Compliance Criteria
Prioritize these factors when selecting equipment:
- Regulatory compliance (21 CFR Part 11 for electronic systems with audit trails and user authentication)
- Measurement range and accuracy (particle counters must meet ISO 21501-4 calibration standards)
- Calibration requirements (annual calibration is mandatory for most instruments)
- Data integrity features including secure storage and automated timestamping
- Integration capabilities with existing facility systems
Install Equipment at Predetermined Locations
When mounting equipment, confirm:
- Proper height for representative sampling
- Accessibility for routine maintenance
- Minimal interference with manufacturing operations
- Adequate protection from temperature or humidity extremes
For continuous monitoring systems in Grade A zones, use sample flow rates of at least 28 liters per minute to capture transient contamination events.
Set up wired or wireless sensor networks connecting to central data servers or cloud-based platforms. Implement real-time dashboards for operations teams and automated alert notification systems (email, SMS, BMS alarms) that trigger when parameters exceed alert limits.
Facilities built with integrated monitoring infrastructure from the start simplify this considerably. ACH Engineering designs cleanrooms with pre-planned equipment mounting points, walkable ceiling systems for maintenance access, and modular layouts that accommodate future monitoring expansion — reducing installation complexity and long-term operational overhead.

Step 3: Validate and Qualify the Environmental Monitoring System
Installation Qualification (IQ)
Verify all equipment is installed per specifications:
- Proper equipment labeling and location documentation
- Correct connection to power and data networks
- Documentation of serial numbers, calibration certificates, and manufacturer specifications
- Verification of tubing lengths for particle counters (typically <1m to prevent particle loss)
Operational Qualification (OQ)
Confirm each monitoring instrument performs within specified ranges:
- Measurement accuracy across the full operating range
- Alarm setpoints trigger appropriately at defined thresholds
- Data is accurately recorded with proper timestamps
- System security controls (user authentication, access restrictions) function as intended
- Flow rates meet specifications (e.g., air samplers, particle counter sample flow)
Performance Qualification (PQ)
Demonstrate the complete EM system meets all acceptance criteria during representative manufacturing operations. PQ typically spans 6-12 months to capture seasonal variations and establish robust baseline data.
Requalification schedules by classification:
| Area Classification | Requalification Frequency |
|---|---|
| Grade A / B | Every 6 months |
| Grade C / D | Every 12 months |

Compile all qualification activities, deviations, and resolutions in formal validation reports reviewed and approved by Quality Assurance. Establish calibration and maintenance schedules per manufacturer recommendations — typically annual for particle counters, air samplers, and environmental sensors.
Step 4: Establish Operational Procedures and Data Management Protocols
Standard Operating Procedures (SOPs)
SOPs must cover all aspects of EM program execution:
- Aseptic sampling techniques for viable monitoring
- Equipment operation and troubleshooting
- Sample handling, incubation conditions, and colony counting
- Data recording requirements and review timelines
- Deviation investigation procedures
- Corrective and preventive action (CAPA) protocols
Data Integrity Controls
All EM data must meet ALCOA+ principles — Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, and Available. In practice, this means:
- Controlled access with unique user IDs
- Automated audit trails capturing all data modifications
- Secure storage with regular backups
- Electronic signatures where required by 21 CFR Part 11
Personnel Training
Provide documented training on SOPs, aseptic technique, equipment operation, data recording, and deviation reporting. Conduct periodic competency assessments to verify ongoing proficiency. FDA warning letters consistently cite personnel behavior as a primary contamination source — a well-documented training program is one of the most defensible investments in your EM program.
Data Review and Trending
Implement multi-level review across four timeframes:
- Daily: Operations reviews monitoring results for immediate excursions
- Weekly: Quality conducts trending analysis identifying patterns
- Monthly: Management reviews EM program effectiveness metrics
- Annually: Comprehensive program review with continuous improvement initiatives

Link the EM program to deviation management, CAPA systems, change control, and periodic review processes within your QMS. This integration keeps compliance continuous and allows systematic program improvements as performance data accumulates over time.
When Should You Implement Environmental Monitoring in Cleanroom Manufacturing?
Environmental monitoring is not optional—it's a regulatory requirement for any facility manufacturing sterile products, aseptically processed pharmaceuticals, biologics, cell and gene therapies, or medical devices where contamination poses patient safety risks.
Specific triggering events requiring EM implementation:
- New cleanroom construction or commissioning before production begins
- Changes to manufacturing processes or product types that alter contamination risk
- Facility expansions, modifications, or reclassification of existing spaces
- Regulatory inspections revealing gaps in current monitoring programs
- Trending data indicating loss of environmental control or increasing excursions
- Introduction of new product lines requiring different cleanliness classifications
Risk-based monitoring requirements:
Monitoring intensity depends directly on classification and process risk. The table below summarizes the core distinction:
| Classification | Zone Type | Monitoring Approach |
|---|---|---|
| ISO 5 / Grade A | Aseptic processing (critical) | Continuous real-time monitoring of viable and non-viable particles during all critical operations |
| ISO 7–8 / Grade C–D | Support areas | Periodic monitoring at validated frequencies based on historical performance; pressure differentials monitored continuously |
ISO 5/Grade A zones require continuous real-time data because transient contamination events—brief spikes that resolve quickly—are invisible to periodic grab samples. Support areas carry lower risk, so periodic monitoring with a strong historical baseline is generally acceptable.
What You Need Before Implementing Environmental Monitoring in Cleanroom Manufacturing
Successful EM implementation requires significant upfront planning, infrastructure readiness, and organizational commitment before collecting the first sample.
Equipment and Technology Requirements
Minimum equipment needed:
- Particle counters: Portable units ($2,000-$15,000) for periodic monitoring or fixed systems for continuous monitoring in critical zones
- Viable air samplers: Active volumetric samplers for quantitative air sampling
- Settle plates and contact plates: With appropriate growth media (Tryptone Soy Agar for bacteria, Sabouraud Dextrose Agar for fungi)
- Environmental sensors: Calibrated temperature and humidity sensors
- Pressure monitors: Differential pressure gauges between adjacent zones
- Data management systems: Manual logbooks (for simple programs) or automated BMS/EMS with electronic data capture
Ongoing costs to budget:
- Annual calibration: Mandatory for all instruments; typically $600-$800 per unit
- Deviation investigations: An out-of-tolerance calibration can trigger investigations costing $8,000-$12,000 in labor and resources — calibration is the cheaper problem to have
Facility Infrastructure and Design Readiness
HVAC system qualification:
Verify that cleanroom HVAC systems are commissioned and qualified before monitoring begins. Key performance parameters to confirm:
- Temperature: 20-24°C (stable, within spec)
- Humidity: 30-60% RH
- Air changes per hour: >20 ACH for ISO 8; >200 ACH for ISO 5/6
- Pressure differentials: ≥10-15 Pa between adjacent zones
Cleanroom construction completion:
Confirm construction is complete and compliant before commencing monitoring. Required features include:
- Non-shedding, cleanable surface finishes throughout
- Adequate lighting to support sampling activities
- Sealed wall-to-ceiling and wall-to-floor transitions (cleanroom coving)
- Equipment access points that don't compromise environmental control
Regulatory and Compliance Foundation
Identify applicable regulatory frameworks:
Determine which standards apply to your operation:
- FDA 21 CFR 210/211 for pharmaceutical manufacturing
- EU GMP Annex 1 (2022) for sterile product manufacturing
- ISO 14644 for cleanroom classification
- USP <797> for pharmaceutical compounding
- USP <1116> for microbiological evaluation of cleanrooms
Obtain current copies of applicable guidance documents to inform program design and ensure your monitoring strategy aligns with regulatory expectations.
Personnel Qualifications and Training Readiness
Ensure qualified personnel availability:
- Microbiologists or quality professionals who can design the program, set alert/action limits, and interpret results
- Trained technicians competent in aseptic sampling technique and equipment operation
- Maintenance staff to handle calibration schedules, repairs, and troubleshooting
Plan comprehensive training program:
Training must be completed before program launch and should cover aseptic technique, sampling procedures, equipment operation, data integrity, and deviation management. Document all training with signed records and schedule periodic competency assessments to maintain proficiency over time.
Key Parameters to Monitor in Cleanroom Environments
Effective EM programs track multiple parameters simultaneously. Requirements vary by cleanroom classification, process type, and applicable regulatory standard — each demanding a different combination of monitoring methods and frequency.
Non-Viable Particulate Monitoring
Non-viable particle monitoring measures airborne particles ≥0.5 µm and ≥5.0 µm using laser particle counters to verify HVAC filtration effectiveness and detect potential contamination sources before they impact product.
Particle count limits by classification:
| ISO Class | EU GMP Grade | ≥0.5 µm (particles/m³) | ≥5.0 µm (particles/m³) |
|---|---|---|---|
| ISO 5 | Grade A | 3,520 | 29 |
| ISO 7 | Grade B (at rest) / Grade C (in operation) | 352,000 | 2,930 |
| ISO 8 | Grade D | 3,520,000 | 29,300 |
Monitoring approach differences:
Grade A/ISO 5 critical zones require continuous real-time monitoring during critical operations with sample flow rates of at least 28 liters per minute. Lower classification support areas may use periodic monitoring with frequencies determined by risk assessment and historical performance data.
Viable Particulate (Microbial) Monitoring
Viable monitoring detects and quantifies living microorganisms — bacteria, yeast, and mold — in air and on surfaces using culture-based methods including settle plates, active air sampling, contact plates, and swabs.
Action limits by cleanroom grade (EU GMP Annex 1):
| Grade | Air Sample (CFU/m³) | Settle Plates (CFU/4h) | Contact Plates (CFU/plate) | Glove Print (CFU/glove) |
|---|---|---|---|---|
| Grade A | No Growth | No Growth | No Growth | No Growth |
| Grade B | 10 | 5 | 5 | 5 |
| Grade C | 100 | 50 | 25 | - |
| Grade D | 200 | 100 | 50 | - |
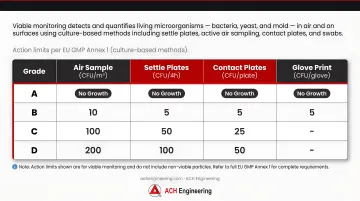
For Grade A zones, any microbial recovery triggers immediate investigation. Use appropriate growth media and incubation conditions:
- Bacteria: Tryptone Soy Agar (TSA), incubated at 30–35°C
- Fungi: Sabouraud Dextrose Agar (SDA), incubated at 20–25°C
- Incubation period: 5–7 days to maximize recovery
Environmental Conditions: Temperature, Humidity, and Pressure
Temperature and humidity control:
Temperature and humidity directly affect both product stability and personnel performance under gowning requirements. Uncontrolled fluctuations risk product degradation or condensation on surfaces. Typical pharmaceutical cleanroom tolerances:
- Temperature: ±2°C from setpoint
- Humidity: ±5% RH from setpoint
Pressure differential monitoring:
Pressure differentials prevent contamination migration between cleanroom zones. Two configurations apply depending on process risk:
- Positive pressure cascade: Air flows from cleaner to less-clean zones, protecting product
- Negative pressure: Contains hazardous materials or viable organisms within a zone
Maintain pressure differentials ≥10–15 Pa between adjacent zones of different classification. Continuous monitoring with automated alarms is required if limits are breached.
Airflow Velocity and Patterns
Unidirectional flow verification:
Airflow velocity monitoring verifies unidirectional (laminar) flow in critical Grade A zones maintains specified velocity — typically 0.36–0.54 m/s (70–110 fpm) — to continuously sweep particles away from exposed product.
Airflow visualization:
Smoke studies or airflow visualization techniques verify proper airflow patterns, absence of turbulence or dead zones, and contamination removal from critical areas during initial qualification and periodic requalification.
Common Mistakes When Implementing Environmental Monitoring in Cleanroom Manufacturing
1. Inadequate Risk Assessment
Insufficient monitoring locations, missed critical control points, or monitoring frequencies that don't reflect actual contamination risk are among the most common — and most cited — regulatory findings. Use your Contamination Control Strategy as the foundation for any risk assessment, not an afterthought.
2. Cost-Driven Equipment Selection
Choosing monitoring equipment based solely on purchase price creates downstream problems. Evaluate each option against:
- Measurement accuracy and instrument precision
- Regulatory compliance requirements (21 CFR Part 11)
- Calibration frequency and associated costs
- Data integrity features and audit trail capability
- Long-term total cost of ownership
Poor equipment choices routinely generate higher operational costs through repeat calibrations, investigations, and regulatory findings that could have been avoided upfront.
3. Inappropriate Alert and Action Limits
Limits set without grounding in qualification data and regulatory guidance will fail in one of two directions: too tight triggers excessive false alarms and investigation burden; too loose misses true excursions before they reach product quality. Neither outcome is acceptable in a regulated facility.
4. Insufficient Personnel Training
Personnel are the primary contamination vector in cleanrooms. When training on aseptic sampling technique, equipment operation, and data integrity falls short, the consequences show up fast — contaminated samples, invalid results, and data integrity violations flagged during inspections. Thorough, consistent training is non-negotiable.
5. Poor QMS Integration
When the environmental monitoring program operates in isolation from the facility's Quality Management System, deviation investigations stall, CAPA implementation lags, and trending opportunities get missed. Regulators look for evidence of continuous improvement — a disconnected EM program makes that case difficult to make.
Troubleshooting Environmental Monitoring Issues in Cleanroom Manufacturing
Even well-designed environmental monitoring programs produce unexpected results. When deviations occur, a structured troubleshooting approach helps isolate the root cause quickly — before a transient excursion becomes a persistent contamination risk.
Problem: Elevated Non-Viable Particle Counts
Common causes:
- HEPA filter degradation or breach (media leaks occur in approximately 1.4% of filters, housing leaks in 0.6%)
- Improper gowning or personnel behavior generating particles
- Equipment malfunction or material shedding
- Facility construction or maintenance activities disrupting airflow
- Particle counter malfunction or calibration drift
Investigation steps:
- Verify particle counter calibration and function before drawing conclusions
- Inspect HEPA filters and room seals for visible damage or bypass leaks
- Review recent maintenance activities or facility changes that may have disrupted airflow
- Observe personnel gowning practices during live operations
- Increase monitoring frequency to determine whether the excursion is persistent or transient

Problem: Microbial Growth Detected in Critical Areas
Common causes:
- Inadequate cleaning and disinfection (wrong agent, insufficient contact time, improper technique)
- Compromised facility integrity (cracks in surfaces, damaged seals, breaches in walls or ceilings)
- Contaminated materials or equipment entering the cleanroom
- Aseptic technique failures by personnel during interventions
Investigation steps:
- Identify the organism through microbial identification testing to determine the source — environmental flora vs. human flora indicates very different origins
- Review cleaning logs and verify disinfectant efficacy and rotation schedules
- Inspect facility surfaces and seals for cracks, damage, or breaches
- Evaluate personnel gowning and aseptic technique through direct observation
- Conduct targeted environmental swabbing to map and isolate the contamination source
Problem: Loss of Pressure Differential Between Zones
Common causes:
- HVAC system malfunction (fan failure, damper positioning issues)
- Blocked or dirty filters restricting airflow
- Doors left open or door seals compromised
- Facility modifications affecting room volume or air balance
Investigation steps:
- Verify HVAC system operation and confirm alarm functionality is active
- Check filter pressure drops across pre-filters and HEPA filters for blockage
- Inspect door seals, gaskets, and interlock systems for wear or compromise
- Review recent facility changes or modifications that may have altered room volume or air balance
- Rebalance airflow if needed and document all corrective actions
Conclusion
Environmental monitoring is a critical quality system ensuring cleanroom manufacturing environments remain under control and compliant with regulatory requirements. A well-executed EM program protects product quality, patient safety, and business continuity — catching contamination issues before they reach the product.
Successful EM programs share five common foundations:
- Careful planning during the risk assessment phase
- Technology selection aligned with regulatory requirements
- Procedures documented in clear, audit-ready SOPs
- Personnel trained in aseptic technique and data integrity
- Continuous data review with timely response to deviations
With regulatory expectations continuing to evolve toward continuous monitoring and stricter contamination control strategies, facilities that invest in thorough EM programs and partner with experienced cleanroom engineering services providers are better equipped to pass audits, reduce batch failures, and maintain uninterrupted operations as standards tighten.
Frequently Asked Questions
What is the difference between viable and non-viable environmental monitoring?
Non-viable monitoring detects all airborne particles (living and non-living) using laser particle counters, providing real-time data on particle concentrations. Viable monitoring specifically detects living microorganisms using culture-based methods like settle plates and air samplers. Both are required for comprehensive cleanroom control—non-viable monitoring verifies filtration effectiveness, while viable monitoring detects biological contamination risks.
How much does it cost to implement an environmental monitoring program?
Costs vary widely based on cleanroom size and classification. Typical investments range from CAD $5,000–$100,000+ for equipment, CAD $10,000–$50,000 for installation and validation, and CAD $5,000–$20,000 annually for consumables, calibration, and personnel time. Facilities should weigh these costs against the financial risk of undetected contamination events.
What qualifications do personnel need to perform environmental monitoring?
EM personnel need documented training in microbiology or quality systems, aseptic technique, gowning protocols, and sampling procedures. Formal education requirements vary by role, but all staff must pass competency assessments before performing monitoring activities independently.
How often should cleanroom environmental monitoring equipment be calibrated?
Most monitoring equipment—including particle counters, air samplers, and temperature/humidity sensors—requires annual calibration. High-use or critical instruments may need more frequent servicing. All calibration must be performed by qualified providers using traceable standards, with certificates kept on file.
What are the most common causes of environmental monitoring excursions?
The most common causes include personnel errors (improper gowning, poor aseptic technique during interventions), inadequate cleaning and disinfection (wrong agents, insufficient contact time), facility issues (HEPA filter breaches, seal failures, compromised surfaces), equipment problems (particle shedding, contaminated materials), and material transfer contamination. Personnel remain the primary contamination vector in most cleanroom environments.
Can environmental monitoring be outsourced or does it need to be done in-house?
Routine sampling and real-time monitoring must be performed by on-site facility personnel, since immediate excursion response requires trained staff on the floor. Support functions like calibration, microbial identification, and data system hosting can be outsourced. A hybrid model—in-house sampling with outsourced support—is the most practical approach for most facilities.


