
Key Takeaways
- cGMP cleanroom solutions providers deliver turnkey controlled environments meeting FDA 21 CFR 210/211, EU GMP Annex 1, and ISO 14644 standards
- Critical selection factors: regulatory track record, modular vs. traditional construction, validation documentation support, material quality, and post-installation service
- Modular construction cuts time-to-market by 40–50%, completing in 9–18 months vs. 15–30 months for traditional builds
- ACH Engineering is an ISPE member specializing in modular pharmaceutical cleanrooms with end-to-end project management across North America
- The right manufacturer partnership supports audit readiness, scalability, and compliance as pharmaceutical regulations evolve
What is a cGMP Cleanroom Manufacturer?
A cGMP cleanroom solutions provider is a specialized company that designs, builds, constructs, and validates controlled environments meeting Current Good Manufacturing Practice standards for pharmaceutical and biotech production.
Their scope extends well beyond construction. These manufacturers provide regulatory consulting, material selection expertise, HVAC system design, environmental monitoring integration, and comprehensive validation documentation support that regulatory submissions rely on.
The industry breaks down into three main manufacturer types:
- Turnkey solution providers — handle design-build projects end-to-end
- Modular cleanroom specialists — focus on prefabricated, reconfigurable systems
- Traditional construction firms — operate dedicated cleanroom divisions within broader construction practices
Top-tier manufacturers hold memberships in bodies like ISPE (International Society for Pharmaceutical Engineering) and demonstrate proven track records with successful Health Canada and FDA inspections.
These manufacturers must navigate multiple regulatory frameworks simultaneously: FDA 21 CFR Parts 210/211, EU GMP Annex 1, ISO 14644 cleanroom classifications, and USP 797/800 standards for pharmaceutical compounding. That regulatory depth is non-negotiable. 22% of FDA warning letters in Fiscal Year 2025 cited violations of 21 CFR 211.42 (Design and Construction Features), with aseptic processing areas flagged most often. Manufacturers without deep compliance knowledge expose clients to serious regulatory risk and potential market access delays.
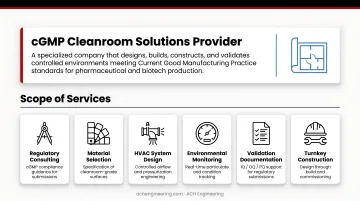
Core Components of cGMP Cleanroom Systems
Procurement teams evaluating cleanroom manufacturers need to assess more than design aesthetics — the underlying technical systems determine whether a facility will pass qualification and sustain compliance. These systems must work in coordination to maintain contamination control across every zone.
Structural Systems and Materials
Pharmaceutical cleanrooms require non-porous, cleanable surfaces that resist microbial growth and withstand aggressive sanitization protocols. Wall, ceiling, and flooring materials typically include epoxy-coated modular panels, coved vinyl flooring, and stainless steel components. These materials must be smooth, impervious, and unbroken to minimize particle shedding and prevent contamination accumulation in corners or seams.
EU GMP Annex 1 (2022) mandates that surfaces permit repeated application of cleaning agents and disinfectants without degradation. Seamless cleanroom coving — which eliminates sharp corners at wall-ceiling and wall-floor junctions — is now a compliance requirement, not just a best practice.
HVAC and Environmental Control
HEPA and ULPA filtration systems form the foundation of contamination control. Key environmental parameters for pharmaceutical cleanrooms include:
| Parameter | Requirement |
|---|---|
| Airflow speed (ISO Class 5 / aseptic zones) | 0.36–0.54 m/s (unidirectional, homogeneous) |
| Pressure differential between adjacent grades | Minimum 10 Pascals (Pa) |
| Temperature range | 18°C to 24°C |
| Relative humidity | 30% to 60% |
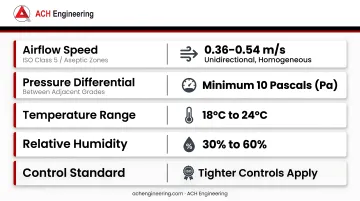
Tighter controls apply when specific process requirements demand enhanced environmental stability beyond these baseline thresholds.
Monitoring and Automation Systems
Integrated Environmental Monitoring Systems (EMS) and Building Management Systems (BMS) provide real-time tracking of temperature, humidity, pressure differentials, and particle counts. These systems must comply with FDA 21 CFR Part 11, ensuring electronic records are secure, time-stamped, and accessible only to authorized personnel.
The 2022 EU GMP Annex 1 revision raised the bar on requalification frequency:
- Grade A and B areas: requalification every 6 months
- Grade C and D areas: requalification every 12 months
Manufacturers must deliver monitoring solutions that support this increased testing cadence without interrupting production schedules.
Benefits of cGMP Cleanroom Manufacturers
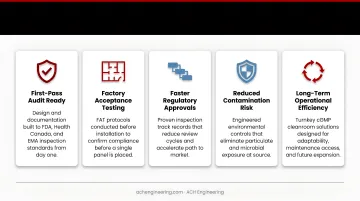
Specialized manufacturers deliver value far beyond basic construction—they enable faster regulatory approvals, reduce contamination risks, and support long-term operational efficiency. For pharmaceutical companies, the difference between an experienced cGMP manufacturer and a general contractor often shows up at the worst possible moment: during an FDA or Health Canada inspection.
- Design to first-pass audit standards, backed by proven FDA/EMA inspection track records
- Conduct Factory Acceptance Testing (FAT) off-site, reducing on-site commissioning time and cost
- Specify materials based on real regulatory inspection outcomes, not just theoretical compliance
- Deliver complete IQ/OQ/PQ documentation packages that satisfy both internal audits and regulatory reviews
- Build modular systems that expand, relocate, or reconfigure without full reconstruction
- Provide ongoing preventive maintenance to keep facilities continuously compliant
- Embed regulatory requirements into the original design, eliminating expensive retrofits later
Industry estimates put pharmaceutical recall costs at $2.5–$5 billion USD annually, with facility design failures among the most preventable root causes. A qualified cGMP manufacturer removes that risk before the first brick is laid—not after a failed inspection forces a costly redesign.
What to Consider When Choosing the Best cGMP Cleanroom Manufacturer
Manufacturer selection directly impacts project timeline, total cost of ownership, regulatory compliance, and operational flexibility for 10–20+ years. The following factors help procurement teams connect manufacturer capabilities to measurable business outcomes like time-to-market, audit pass rates, and lifecycle costs.
Regulatory Expertise and Track Record
Your manufacturer must demonstrate deep knowledge of FDA 21 CFR 210/211, EU GMP Annex 1, ISO 14644 standards, and USP requirements — backed by a proven history of successful regulatory inspections. Request specific examples of facilities that have passed FDA or EMA audits, including inspection dates and outcomes.
22% of recent FDA warning letters cited design deficiencies — meaning regulatory expertise directly affects your inspection readiness. This factor shapes approval timelines, audit pass rates, compliance risk, and your ability to support multi-market launches.
Modular vs Traditional Construction Approach
This decision fundamentally affects project economics and flexibility. Modular systems offer 9–18 month deployment versus 15–30 months for traditional construction — approximately 40–50% faster. Modular construction provides factory-controlled quality, easier validation through off-site FAT, and future scalability. Traditional construction offers site-specific customization but longer timelines and higher on-site labor costs.
Financial advantage: modular assets often qualify for 7-year accelerated depreciation (classified as equipment) versus 39 years for traditional structures (classified as real property), delivering significant cash flow benefits.
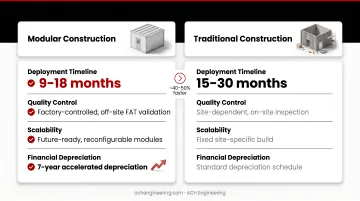
This choice directly influences time-to-production, capital efficiency, and your ability to relocate or expand without major disruption.
Validation and Documentation Support
Comprehensive validation services are essential. Your manufacturer should provide or directly support Installation Qualification (IQ), Operational Qualification (OQ), and Performance Qualification (PQ) protocols with complete documentation packages ready for regulatory inspection.
Manufacturers who perform Factory Acceptance Testing (FAT) prior to delivery cut on-site commissioning time and reduce validation complexity considerably. Key impacts include:
- Shorter commissioning timelines
- Stronger inspection readiness from day one
- Reduced long-term compliance maintenance burden
Material Quality and Cleanability Standards
All surfaces must be non-porous, non-shedding, chemically resistant, and designed for repeated sanitization without degradation. Material selection directly impacts contamination control effectiveness and long-term facility performance.
Wall panels, flooring, and ceiling systems must meet EU GMP Annex 1 requirements for smooth, impervious surfaces — and seamless coving at all junctions is now a regulatory requirement, not a design preference. Material choices drive contamination control, cleaning validation outcomes, microbial control, and long-term replacement costs.
Technical Capabilities and System Integration
Your manufacturer should handle the full technical stack as integrated systems rather than outsourcing pieces to separate vendors. That means ownership of:
- HVAC design and pressure cascade engineering
- Environmental monitoring integration
- Utility coordination across all building systems
Verify they can guarantee 10 Pa pressure differentials between adjacent zones, maintain ISO Class 5/7/8 classifications under all operating conditions, and provide HEPA integrity testing at least annually.
Single-source technical accountability improves system reliability, simplifies maintenance, and reduces the risk of gaps between vendors during commissioning or deviations.
Post-Installation Support and Service Network
Cleanrooms require preventive maintenance, calibration, filter replacements, system upgrades, and rapid response to deviations or failures. Production halts cost millions in lost revenue — your manufacturer's service network directly impacts business continuity.
When evaluating service support, prioritize:
- Response time commitments for critical failures
- Spare parts availability and lead times
- Local technical presence versus remote-only coverage
For North American operations, ACH Engineering's regional coverage from Ontario to Alberta — and across the broader US market — means faster on-site response than international-only vendors. That difference in response time can be the difference between a minor deviation and a costly production halt.
How ACH Can Help
ACH Engineering is a turnkey controlled environment solutions provider built around pharma, biotech, and compounding pharmacy applications. The company holds memberships with ISPE, PEO, and PMI—credentials that reflect both pharmaceutical engineering knowledge and rigorous project management standards. Service coverage runs from Ontario to Alberta and across the USA, with a Calgary office supporting western Canadian clients.
ACH Engineering's key differentiators for pharmaceutical applications include:
- Modular construction that completes installation in weeks rather than months, reducing costs versus site-built alternatives
- Tailored designs for compounding pharmacies (USP 797/800), pharmaceutical manufacturing (cGMP/FDA), and biotech environments
- Full project management from design through validation support, covering material sourcing, vendor coordination, and construction oversight
- Adaptable systems that accommodate future expansion, relocation, or process changes without full reconstruction
- Prefabricated components and a streamlined design-build process that compress time-to-production
- ISO Class 1–9 compliance capabilities across North American regulatory frameworks (FDA, cGMP, USP)

For pharmaceutical teams navigating tight timelines or facility upgrades, ACH's design-build model offers a direct path from process requirements to a validated, compliant cleanroom. Contact ACH at sales@achengineering.com or +1 647-406-5721 to discuss your project scope.
Conclusion
Selecting the right cGMP cleanroom solutions provider is a strategic decision that impacts regulatory success, operational efficiency, and long-term competitiveness. With pharmaceutical recalls costing up to $5 billion annually and regulatory citations for design deficiencies increasing, the cost of choosing the wrong partner is measurable — in both dollars and compliance risk.
The goal is finding a manufacturer whose capabilities, approach, and support model align with your specific production requirements, regulatory obligations, and growth trajectory — not simply the lowest bid. Evaluate candidates across six areas:
- Regulatory track record and documentation history
- Construction methodology and cleanroom classification experience
- Validation support (IQ/OQ/PQ protocols and reporting)
- Material quality and supply chain reliability
- Technical integration (HVAC, BMS, monitoring systems)
- Post-installation service and response commitments
As pharmaceutical regulations evolve and advanced therapies demand more sophisticated manufacturing environments, the solutions provider you choose will either accelerate or complicate every regulatory milestone ahead. ACH Engineering brings end-to-end project management, ISPE membership, and tailored controlled environment solutions across North America — the kind of structured support that keeps facilities audit-ready and operationally sound long after installation is complete.
Frequently Asked Questions
What is the difference between GMP and cGMP in cleanroom manufacturing?
GMP (Good Manufacturing Practice) refers to general quality standards, while cGMP (current Good Manufacturing Practice) emphasizes that manufacturers must use current technologies and approaches to meet evolving regulatory expectations. The "c" in cGMP reminds manufacturers that compliance is not static—systems must stay up-to-date with current standards as technology and science advance.
How long does it take to design and install a cGMP cleanroom for pharmaceutical production?
Modular cleanroom solutions typically reach operational status in 9-18 months, compared to 15-30 months for traditional construction — a 40-50% reduction through parallel off-site fabrication. Final timelines depend on complexity, ISO classification, size, and regulatory validation requirements.
What ISO cleanroom classification is required for pharmaceutical manufacturing?
Most pharmaceutical production uses ISO Class 7 or 8 (EU Grade C/D) for general manufacturing areas. ISO Class 5 (EU Grade A) is required for aseptic processing and sterile product filling operations, with unidirectional airflow providing 0.36-0.54 m/s air speed at the working position.
Do cGMP cleanroom solutions providers offer validation documentation?
Yes — qualified manufacturers deliver full validation packages covering Installation Qualification (IQ), Operational Qualification (OQ), and Performance Qualification (PQ) for regulatory inspections. Prioritize manufacturers who conduct Factory Acceptance Testing (FAT) off-site to accelerate on-site commissioning.
What are the cost differences between modular and traditional cGMP cleanrooms?
While initial material costs may be similar, modular cleanrooms typically reduce total project costs by 15-30% through faster installation, reduced on-site labor, shorter facility downtime, and accelerated time-to-production. Additionally, modular systems qualify for 7-year accelerated depreciation versus 39 years for traditional construction, providing significant cash flow advantages.
How often do cGMP cleanrooms require recertification and maintenance?
EU GMP Annex 1 (2022) and ISO 14644-2 require requalification every 6 months for Grade A/B areas and every 12 months for Grade C/D. Ongoing maintenance includes annual HEPA filter integrity testing, environmental monitoring calibration, and routine HVAC inspections.


