
Introduction
Cell therapies cannot be terminally sterilized — every contamination event is a product loss event. According to FDA guidance on aseptic processing, any manual or mechanical manipulation of sterile components prior to assembly carries significant contamination risk. In cell therapy, that risk is compounded: living biological material actively supports microbial growth.
For cell therapy manufacturers, cleanroom standards determine whether your facility earns regulatory approval, sustains product safety, and operates at all. Three frameworks govern the landscape: ISO 14644 (technical classification), FDA 21 CFR Part 211 (US cGMP requirements), and EU GMP Annex 1 (European sterile manufacturing). Each covers distinct aspects of cleanroom design, operation, and validation that impact your path to compliant manufacturing.
This guide breaks down the classification requirements, implementation pathways, and compliance strategies you need to design facilities, secure regulatory approval, and maintain validated manufacturing operations.
Key Takeaways
- Three frameworks govern cell therapy cleanrooms: ISO 14644 (particle classification), FDA cGMP (US regulations), and EU GMP Annex 1 (European sterile manufacturing)
- Classifications range from ISO 5/Grade A (critical aseptic steps) to ISO 8/Grade D (support areas), each with specific particle limits and operational controls
- Controlled parameters span airborne particles, air changes per hour, pressure differentials (10–15 Pa), temperature, and humidity
- Compliance demands IQ/OQ/PQ (installation, operational, and performance qualification), ongoing environmental monitoring, and re-certification every 6–12 months
What Cleanroom Standards Represent in Cell Therapy Manufacturing
Cleanroom standards establish the regulatory and technical frameworks that define controlled environment requirements, minimizing contamination risks during cell therapy production. Unlike traditional pharmaceuticals that undergo terminal sterilization, cell therapies involve living material that cannot be heat-treated or radiation-sterilized after processing.
These standards function on two levels:
Design specifications define physical parameters:
- Air quality and particle concentration limits
- Pressure cascade requirements between adjacent spaces
- HVAC system performance criteria
- Surface finishes and materials
Operational controls govern ongoing compliance:
- Environmental monitoring protocols
- Testing frequencies and acceptance criteria
- Maintenance and re-qualification schedules
- Personnel gowning and behavior requirements
How Standards Relate to Each Other
ISO 14644 provides the technical classification system based on airborne particle concentrations. The standard defines nine classes (ISO 1-9), with lower numbers indicating stricter cleanliness requirements. Cell therapy manufacturing typically uses ISO Class 5 through 8.
FDA cGMP and EU GMP set the regulatory requirements for pharmaceutical manufacturing. They reference ISO classifications but add requirements specific to sterile drug production: personnel training, batch documentation, validation protocols, and quality systems.
Together, these systems work in a hierarchy: ISO defines how clean the air must be, while GMP frameworks define how you prove and maintain that cleanliness for regulatory compliance.
Why Cell Therapy Demands Stricter Controls
Cell therapy manufacturing requires more stringent controls than traditional pharmaceuticals for four reasons:
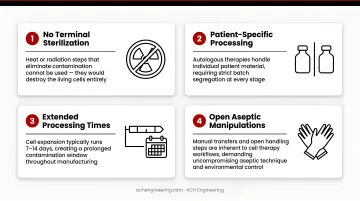
- No terminal sterilization — processing cannot include heat or radiation steps that would kill the cells
- Patient-specific processing — autologous therapies handle individual patient material, requiring strict segregation between batches
- Extended processing times — cell expansion typically runs 7–14 days, creating a prolonged contamination window
- Open aseptic manipulations — harvesting and formulation expose the product directly, with no closed-system protection
Standards must address the complete workflow: starting material receipt, cell isolation, genetic modification (for CAR-T), expansion in bioreactors, formulation, fill-finish, and cryopreservation. Each stage carries its own contamination risks and requires specific environmental controls.
Key Regulatory Frameworks and Their Requirements
Three primary frameworks govern cell therapy cleanroom design and operation. Understanding how ISO standards, FDA cGMP, and EU GMP Annex 1 each define requirements—and where they diverge—is essential before committing to a facility layout or monitoring strategy.
ISO 14644 Standard
ISO 14644-1:2015 defines cleanroom classification based on maximum allowable airborne particle concentrations per cubic meter for particles ≥0.5 μm. The standard establishes nine classes (ISO 1-9), with cell therapy manufacturing utilizing ISO 5-8.
ISO 14644-2 (updated 2015) specifies monitoring requirements, testing frequencies, and performance qualification protocols. It mandates:
- Re-qualification every 6 months for ISO Class 5 environments
- Re-qualification every 12 months for ISO Class 7-8 environments
- Continuous particle monitoring during critical operations
The standard provides the technical measurement framework. It does not specify which classification applies to each manufacturing step. That guidance comes from GMP regulations.
FDA Current Good Manufacturing Practice (cGMP)
FDA 21 CFR Part 211 establishes requirements for facilities and equipment used in pharmaceutical manufacturing. Section 211.42 specifically addresses design and construction features for aseptic processing.
The FDA's 2004 guidance on aseptic processing defines the "Critical Area" (equivalent to ISO Class 5) where sterile products, containers, and closures are exposed to environmental conditions. Key requirements include:
- HEPA-filtered air under positive pressure
- Unidirectional airflow in critical zones
- Pressure differentials of 10-15 Pa between adjacent areas
- Validation through media fills (process simulations)
FDA's 2020 guidance for gene therapy products acknowledges that cell therapies often require release before final sterility results are available (due to short shelf life), allowing conditional release based on in-process testing and rapid microbial detection methods.
EU GMP Annex 1
The 2022 revision of EU GMP Annex 1 introduced sweeping changes for sterile manufacturing. The EU uses a four-grade classification system (A, B, C, D) that maps to ISO classes but adds operational requirements.
Key 2022 updates include:
- A mandatory Contamination Control Strategy (CCS) document covering all critical control points, monitoring locations, and risk mitigation measures across the facility
- Continuous viable air monitoring in Grade A zones during critical processing — periodic sampling alone no longer satisfies the requirement
- Minimum 10 Pa pressure differential between grades, with continuous monitoring and automated alarming required
The revision emphasizes quality risk management and encourages closed systems and isolators to reduce contamination risk.
Comparison and Integration
With all three frameworks in view, the table below shows how their classification systems align — and where the requirements diverge for facilities targeting both markets.
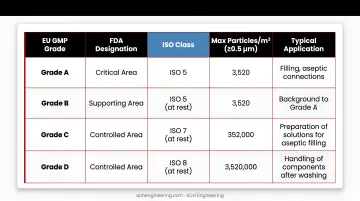
Classification Mapping:
| EU GMP Grade | FDA Designation | ISO Class | Max Particles/m³ (≥0.5 μm) | Typical Application |
|---|---|---|---|---|
| Grade A | Critical Area | ISO 5 | 3,520 | Filling, aseptic connections |
| Grade B | Supporting Area | ISO 5 (at rest) | 3,520 | Background for Grade A |
| Grade C | Class 10,000 | ISO 7 | 352,000 | Solution preparation, early processing |
| Grade D | Class 100,000 | ISO 8 | 3,520,000 | Component handling post-washing |
Facilities targeting both US and EU markets must meet the stricter EU requirements, particularly on continuous monitoring and documented CCS. The 2022 Annex 1 revision is more prescriptive than FDA guidance overall — FDA takes a risk-based approach, while EU Annex 1 specifies exact controls. Designing to EU standards from the outset avoids costly retrofits when seeking dual-market approval.
Classification Levels and Their Applications in Cell Therapy
Different manufacturing stages require different classifications based on contamination risk and product exposure. Understanding which classification applies to each process step is essential for facility design and cost management.
Grade A / ISO Class 5 Applications
Critical aseptic operations requiring Grade A/ISO 5:
- Final formulation and filling operations
- Making sterile connections after filtration
- Opening sterile containers for product transfer
- Critical reagent preparation where sterility cannot be assured by filtration
Design parameters for Grade A zones:
- HEPA-filtered unidirectional airflow at 0.36-0.54 m/s velocity (per EU GMP Annex 1)
- Maximum 3,520 particles/m³ (≥0.5 μm)
- Continuous particle monitoring during operations
- Air change rates exceeding 240-360 ACH (industry practice)
Grade B / ISO Class 7 (at rest) Background Environment
Grade A zones typically operate within Grade B background areas, providing a protective buffer against contamination. The "at rest" designation means Grade B areas must meet ISO 5 particle limits when unoccupied but may reach ISO 7 during operations.
Applications:
- Immediate areas surrounding aseptic operations
- Preparation rooms for Grade A activities
- Gowning areas adjacent to critical zones
- Minimum 10 Pa pressure differential between Grade A and Grade B areas, with continuous monitoring
Grade C / ISO Class 7-8 Support Areas
Applications for less critical steps:
- Early-stage cell processing (initial isolation, washing)
- Non-sterile solution preparation
- Equipment staging and preparation
- Secondary gowning areas
Typical air change rates:
- ISO 7: 40-60 ACH
- ISO 8: 20-40 ACH
Grade C environments balance contamination control with operational flexibility and cost. Many cell separation and early expansion steps can be performed here if subsequent processing includes sterile filtration — or if the process runs in a closed system. That second option, closed systems, carries its own classification implications covered below.
Grade D / ISO Class 8 and Unclassified Areas
Lowest classified areas:
- Material receipt and inspection
- Warehouse storage of components
- Equipment cleaning and staging
- Administrative spaces adjacent to manufacturing
Pressure cascade concept: Facilities must maintain positive pressure differentials from higher to lower classified areas. A typical cascade might be:
- Grade A: +25 Pa (relative to atmospheric)
- Grade B: +15 Pa
- Grade C: +10 Pa
- Grade D: +5 Pa
- Unclassified corridor: 0 Pa (atmospheric)

When this cascade breaks down — due to HVAC failure or improper door sequencing — contamination can travel upstream into critical zones, making continuous pressure monitoring a non-negotiable design requirement.
Closed Systems Reduce Classification Requirements
Both FDA CAR-T guidance and EU GMP Annex 1 encourage closed systems. Operations performed in validated closed isolators can be conducted in Grade D backgrounds rather than Grade B, cutting the infrastructure cost of a Grade B suite — lower HVAC complexity, reduced gowning requirements, and smaller classified footprint overall.
How Standards Are Implemented and Validated
Implementation follows a structured lifecycle from initial design through ongoing operations, with validation at each phase.
Design and Construction Phase
Facility design must document compliance strategies before construction begins:
Required documentation:
- Room layout drawings showing classification zones
- HVAC system specifications (air change rates, filter specifications, pressure cascades)
- Material selections for walls, floors, ceilings (cleanable, non-shedding)
- Pressure cascade plans with setpoints and alarm thresholds
- Equipment placement and workflow diagrams
ACH Engineering's modular cleanroom approach addresses these requirements through prefabricated components (walls, walkable ceilings, flush-design doors) that meet cGMP requirements out of the box. This cuts installation time compared to traditional construction and keeps the facility reconfigurable as processes change or scale.
Commissioning and Qualification (CQV)
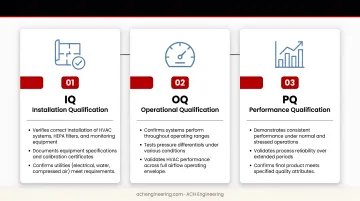
The three-stage qualification process validates that facilities meet design specifications:
Installation Qualification (IQ):
- Verifies correct installation of HVAC systems, HEPA filters, and monitoring equipment
- Documents equipment specifications and calibration certificates
- Confirms utilities (electrical, water, compressed air) meet requirements
Operational Qualification (OQ):
- Confirms systems perform throughout operating ranges
- Tests pressure differentials under various conditions
- Validates air velocity and uniformity in Grade A zones
- Verifies temperature and humidity control
- Conducts HEPA filter integrity testing (PAO/DOP aerosol challenge)
Performance Qualification (PQ):
- Validates sustained compliance under actual operating conditions
- Includes "in operation" particle counts with personnel present
- Performs recovery testing (time to return to specification after disruption)
- Conducts smoke studies to visualize airflow patterns
- May include media fills to validate aseptic technique
Environmental Monitoring Program (EMP)
Ongoing compliance requires continuous or periodic monitoring of viable (microbial) and non-viable (particulate) contamination. Monitoring frequency increases with classification level:
Grade A requirements:
- Continuous particle monitoring during critical operations
- Active air sampling for viable organisms
- Settle plates throughout operations
- Surface sampling after completion
Grades B-D requirements:
- Periodic particle monitoring (frequency based on risk assessment)
- Routine viable monitoring per shift or batch
- Trending and investigation of excursions
The 2022 EU GMP Annex 1 requires that monitoring strategies be defined in the Contamination Control Strategy, with locations and frequencies justified by risk assessment.
Re-Certification Requirements
EMP data informs re-qualification timing, but scheduled re-certification is mandatory regardless of monitoring results. Classification degrades without it:
- Grade A & B (ISO 5): Re-certification every 6 months
- Grade C & D (ISO 7-8): Re-certification every 12 months
- HEPA filter integrity testing: Annually or after any maintenance
- Pressure differential verification: Continuous monitoring with periodic calibration
A rolling re-qualification calendar — staggering room certifications across the year — prevents operational disruptions and keeps all zones within active compliance windows.
Common Compliance Challenges and Misinterpretations
Misconception: Initial Qualification Ensures Permanent Compliance
Achieving classification during commissioning does not guarantee ongoing compliance. Classification degrades due to:
- HEPA filter loading and seal degradation
- HVAC system wear and performance drift
- Changes in room occupancy or equipment
- Facility modifications or expansions
Robust preventive maintenance programs, continuous monitoring of critical parameters, and scheduled re-qualification testing are all required to stay compliant long-term.
Challenge: Applying Standards to Personalized Therapies
Standards designed for traditional pharmaceutical batch manufacturing weren't designed for patient-specific cell therapy workflows — small batch sizes and rapid turnaround requirements create real friction with conventional rules.
Three issues come up repeatedly:
- 14-day sterility tests are too slow for products with 72-hour shelf lives
- Batch-based validation doesn't align with continuous manufacturing models
- Segregation requirements for autologous products quickly strain limited facility capacity
U.S. FDA guidance allows conditional release based on in-process sterility testing (48-72 hours pre-harvest) and rapid microbial detection (Gram staining). Final sterility results are reviewed post-administration, with investigations triggered by any positive findings.
Risk: Over-Classification of Entire Facilities
Some manufacturers classify entire facilities to Grade B or C when risk-based approaches could allow targeted Grade A zones within Grade D support areas.
Over-classifying creates unnecessary construction costs, inflated operational expenses (gowning, monitoring, maintenance), and reduced flexibility for non-critical support functions — none of which improve product safety.
The practical fix is closed systems: isolators and closed bioreactors can enable Grade A operations within Grade D backgrounds. High-grade zones should be targeted only where product exposure actually occurs. Modular construction makes this easier, allowing distinct classification areas to be built within larger support spaces without committing the entire facility to premium-grade requirements.
Conclusion
Cleanroom standards are not static checklists but dynamic frameworks requiring ongoing management, validation, and adaptation. Successful cell therapy manufacturing depends on understanding three layers:
- Technical classifications (ISO 14644) that define particle limits and testing methods
- Regulatory requirements (FDA cGMP, EU GMP) that establish validation, monitoring, and documentation expectations
- Process-specific risks unique to your cell therapy manufacturing workflow
Addressing all three layers becomes more manageable than it might appear: the convergence of ISO, FDA, and EU standards means a properly designed classification strategy can satisfy all three frameworks simultaneously. The 2022 EU GMP Annex 1 revision's emphasis on Contamination Control Strategy and risk-based approaches aligns closely with FDA's flexible guidance, giving manufacturers real room to optimize facility design without sacrificing compliance.
Modular construction approaches — the kind ACH Engineering designs for pharmaceutical and biotech cleanroom facilities across North America — make this optimization practical. Targeted classification zones, capacity expansion, and future process changes can all be accommodated without tearing down and rebuilding, which matters when product timelines and regulatory windows don't wait.
Frequently Asked Questions
What is the difference between ISO cleanroom classifications and GMP grades?
ISO uses numerical classes (5-8) based purely on airborne particle concentrations measured per cubic meter. GMP frameworks (FDA, EU) use letter grades (A-D in EU system) that incorporate both particle limits and operational requirements including personnel behavior, monitoring frequencies, and validation protocols. Grade A corresponds to ISO 5, Grade D to ISO 8.
Which cleanroom classification is required for CAR-T cell manufacturing?
Critical steps—T-cell activation, viral transduction, final formulation, and filling—require Grade A/ISO 5 conditions. Earlier processing steps like cell separation and expansion may be performed in Grade B or C environments if conducted in closed systems. FDA CAR-T guidance recommends closed systems to minimize contamination risk and reduce background classification requirements.
How often must cleanroom classifications be tested and re-certified?
Initial certification occurs after construction during commissioning. Periodic re-certification follows ISO 14644-2 requirements: every 6 months for Grade A/B (ISO 5) and every 12 months for Grade C/D (ISO 7-8). Grade A areas also require continuous particle monitoring during critical operations, not just periodic testing.
Can you start with a lower classification and upgrade later as you scale?
Technically possible but not recommended. Upgrading requires significant HVAC modifications, extensive re-validation, and costly downtime. Design for your target classification from the start—retrofitting later is far more disruptive and expensive than building correctly the first time.
What are the main differences between FDA and EU GMP requirements for cell therapy cleanrooms?
EU GMP Annex 1 is generally more prescriptive, mandating specific air velocities (0.36-0.54 m/s for Grade A) and continuous monitoring requirements. FDA emphasizes risk-based approaches and outcome-focused compliance, allowing more flexibility in how you achieve Class 100 conditions. Facilities targeting both markets must meet the stricter EU requirements, including the documented Contamination Control Strategy introduced in the 2022 Annex 1 revision.
Do autologous and allogeneic cell therapies require different cleanroom standards?
Classification requirements are based on contamination risk and process steps, not therapy type—both require Grade A conditions for critical aseptic operations. Autologous therapies do require additional segregation to prevent cross-contamination between patient-specific batches, achieved through dedicated suites, closed systems, or validated cleaning procedures rather than stricter classifications.


